
© SEQme s.r.o., 2012 - 2024. Všechna práva vyhrazena.
Právní upozornění.
webdesign Beneš & Michl
Technologie next-generation sekvenování produkují v porovnání se Sangerovou technologií obrovská množství dat. Jejich objem pochopitelně závisí na designu experimentu, primárně však na výstupní kapacitě přístroje. V principu je však vždy potřeba vypořádat se s převedením velkého množství dat do takové podoby, kdy je možné s nimi smysluplně pracovat, aby byla možná hlubší analýza získaných sekvencí, která je cílem celého experimentu.
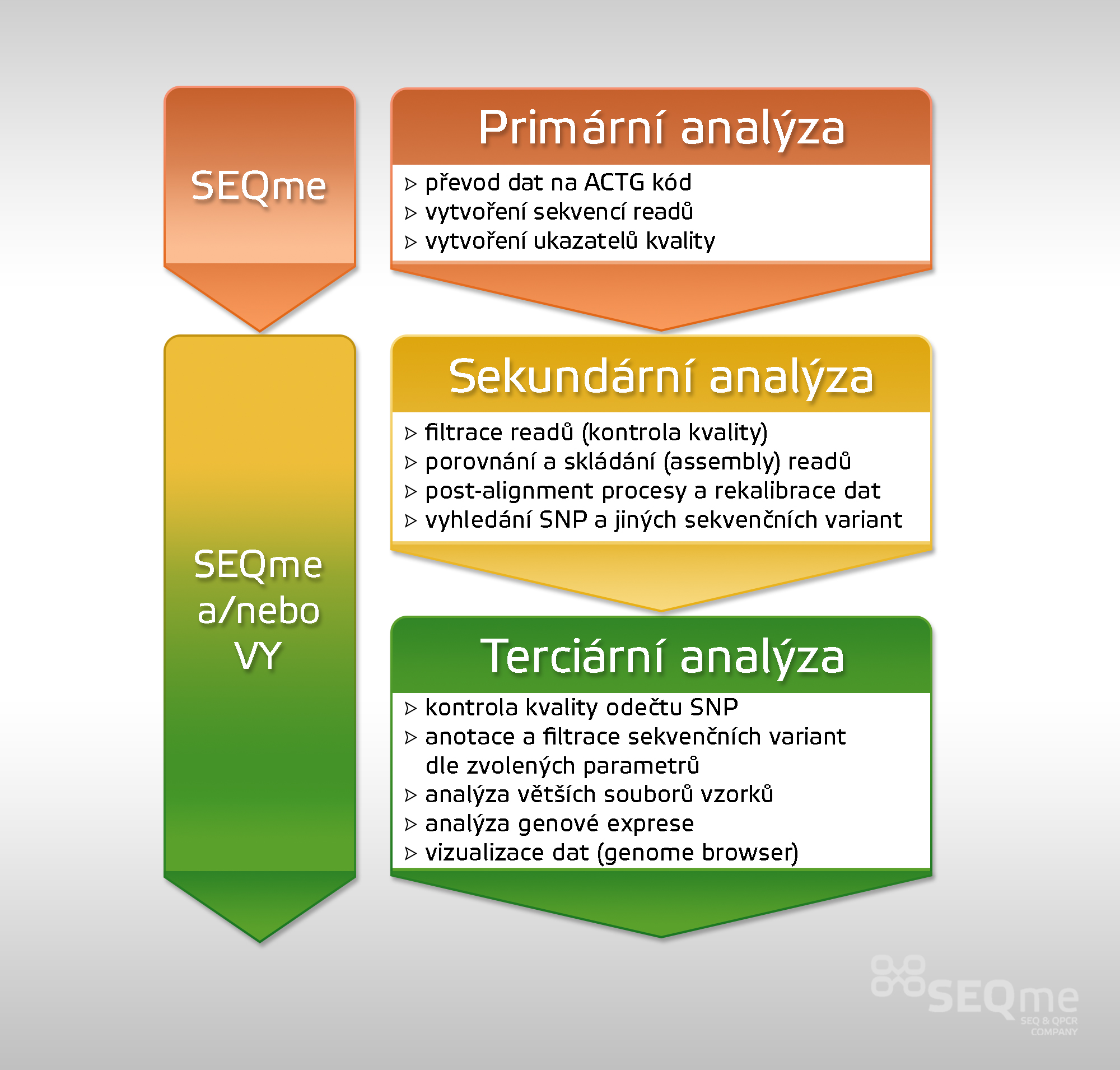
Analýzu dat lze formálně rozdělit do tří úrovní:
Primární analýza
Primární analýzou se rozumí kroky, které vedou k převedení signálu generovaného sekvenátorem na kód ACTG a k výpočtu ukazatelů kvality pro tato data. Tyto kroky jsou pro jednotlivé přístroje specifické. Výstupem je většinou FASTQ soubor v kombinaci s informací o kvalitě dat (Phred, v zásadě jistota správného čtení) pro každou bázi. Primární analýzu provádí sekvenátor a nelze do ní uživatelsky zasáhnout.
Sekundární analýza
Po získání hrubých dat z přístroje (viz primární analýza) je dalším krokem porovnání získaných sekvencí fragmentů („alignment readů“) a jejich spojování v delší sekvence na základě překryvů („assembly readů do kontigů a scaffold“). Těmto krokům ještě předchází filtrace dat tak, aby v sekundární analýze byla použita data s dostatečnou kvalitou.
Proces sekundární analýzy je mnohem méně náročný při použití referenční sekvence, kdy jsou „ready“ skládány nejen na základě vzájemných přesahů, ale také podle shody s referenční sekvencí s přihlédnutím k možným rozdílům (sekvenční variabilita), jejichž analýza je typicky cílem experimentu.
Dalším krokem sekundární analýzy (někdy uváděno až jako terciární) je právě detekce variant. Jedná se o vyhledání odchylek mezi získanou a referenční sekvencí. Tyto odchylky mohou být na úrovni jednotlivých nukleotidů (substituce, kratší inzerce a delece, tzv. "InDels"), nebo se jedná o rozsáhlejší strukturní změny (transverze, translokace, "copy-number" varianty).
Sekundární analýzu lze provádět pomocí vhodných algoritmů a bioinformatických nástrojů, které jsou na jednu stranu předmětem neustálého vývoje, ale na druhou stranu jsou používány již tak dlouho, že dozrály do stádia, kdy se jejich použití standardizuje. Například pro de-novo sekvenování lze využít nástroj Velvet (EMBL-EBI), využívající de Bruijnovy grafy pro budování (assembly) genomické sekvence. Je-li k dispozici referenční sekvence, využívají se např. algoritmy odvozené od Burrows-Wheeler algoritmu (BWA), které dosahují vysoké vyrovnanosti co do rychlosti a přesnosti.
Tyto a další metody sekundární analýzy jsou často uspořádány do „kaskády“ používané pro jeden konkrétní typ analýzy, přičemž ovšem ponechávají možnost variabilního uspořádání a ladění pro další aplikace. Celý proces může být zcela automatizován nebo jej lze naopak měnit a upravovat. Většina těchto metod je volně přístupná a lze je aplikovat s určitou poměrně pokročilou znalostí bioinformatiky a práce s příkazovými řádky nebo pomocí volně dostupných serverových aplikací. Komplexním řešením pak jsou komerční softwarové balíčky různých výrobců cílící na sekundární a/nebo terciární analýzu (CLC bio, Partek, apod.).
Sekundární analýzu provádí sekvenační laboratoř, tedy my, nebo vy, podle dohody v rámci konkrétního projektu. Jejím výstupem jsou typicky soubory ve standardizovaných formátech BAM a VCF, kde BAM je v zásadě alignment readů vůči referenční sekvenci a VCF obsahuje informace o detekovaných variantách.
Terciární analýza
Terciární analýza je poslední fází procesu analýzy dat a poskytuje výstupy, na jejichž základě lze činit vědecké závěry, vyplývající z daného experimentu. Primární a sekundární analýza jsou nezbytným předpokladem tohoto posledního kroku, ale samy o sobě jsou z hlediska interpretace výsledků experimentu vlastně „nezajímavé“.
Terciární analýza je velmi variabilní podle toho, o jakou studii se jedná. V zásadě jde především o hlubší analýzu získaných sekvencí a porovnávání výsledků více vzorků. Proces obvykle nebývá automatizovaný a standardizovaný, jelikož se zpracování liší dle aplikace. Například de-novo sekvenování bude mít zcela jiné zpracování, než resekvenační studie, kde hledáme odchylky a varianty od referenční sekvence. Pro úspěšnou terciární analýzu je samozřejmě klíčový dobře promyšlený design experimentu (počty vzorků, pozitivní a negativní kontroly, replikáty, hloubka čtení atp.).
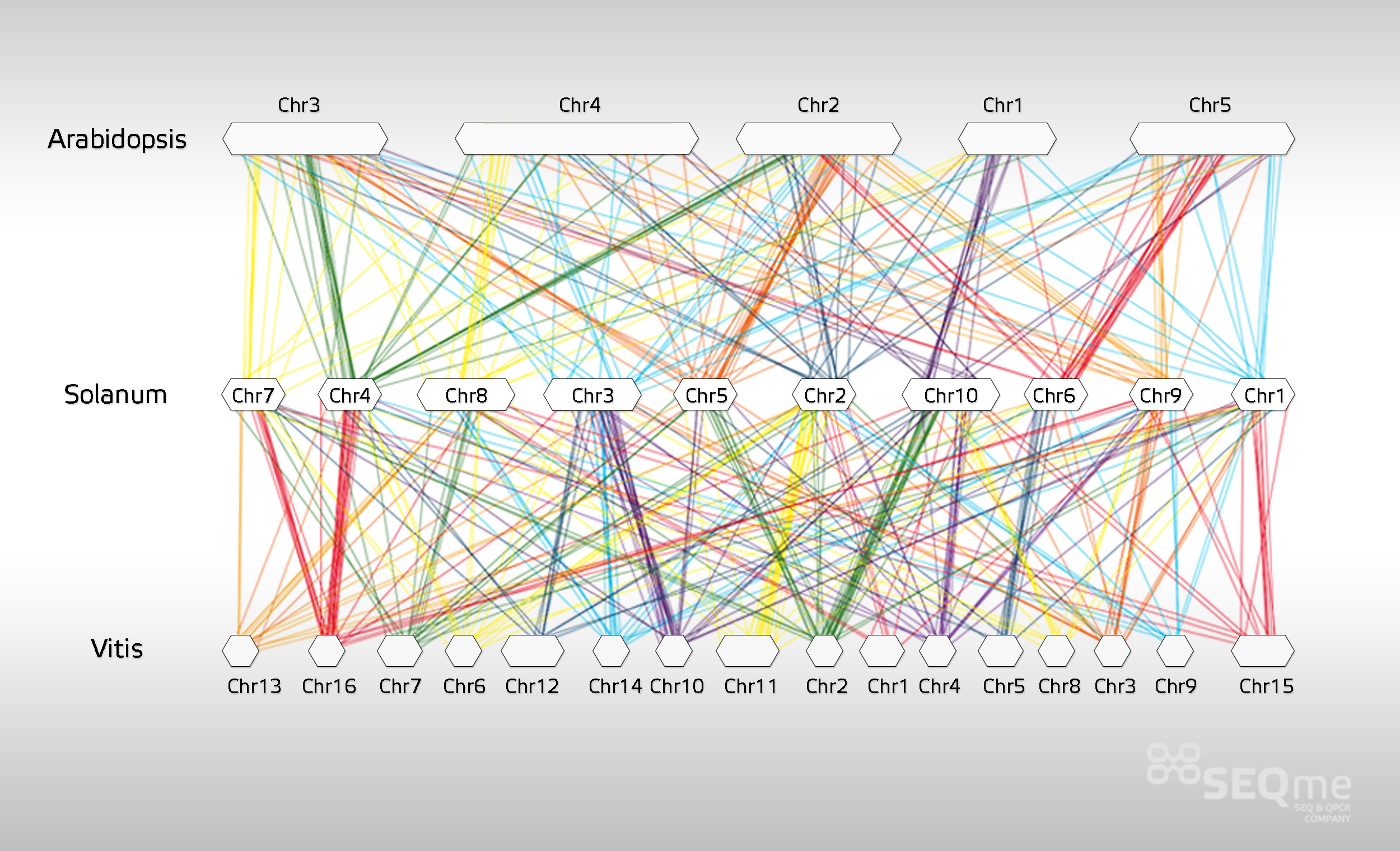
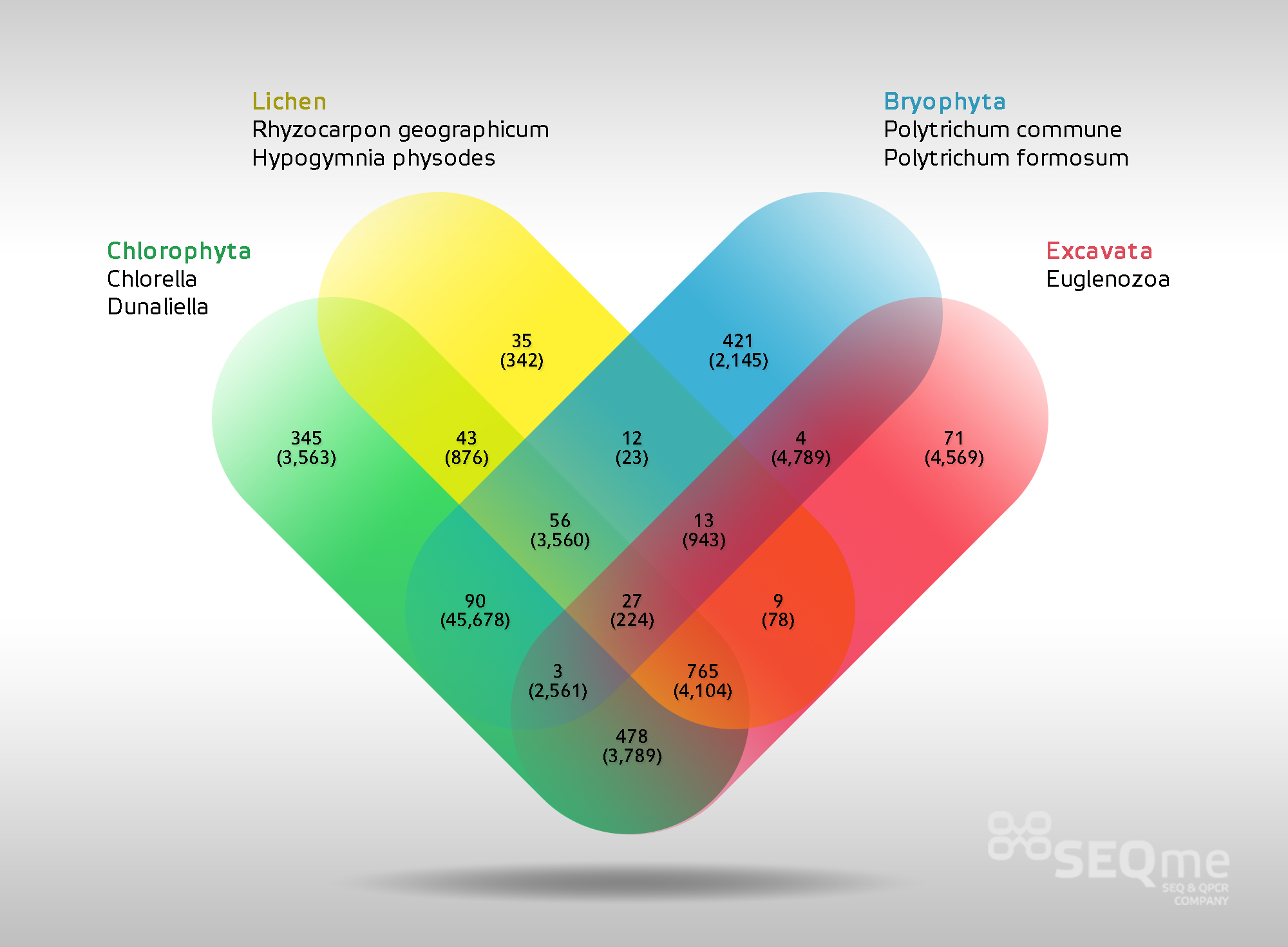
Ukázka výstupů terciární analýzy
Pro provedení terciární analýzy je dále potřeba vhodný software s dobrým uživatelským rozhraním, který umožní práci s výsledky a jejich vizualizaci. Vstupní data jsou typicky ve formátu BAM a VCF. Terciární analýza již není tak hardwarově náročný proces jako předchozí fáze, takže i např. výsledky sekvenace celých genomů lze dále zpracovávat na běžném lehce nadstandardním počítači. Data získaná analýzou exomů nebo cílenou resekvenací jsou ještě méně náročná.
Jako příklad terciární analýzy lze uvést analýzu vzácných sekvenčních variant. Prvním krokem je kategorizace sekvenčních variant s nízkou a vzácnou frekvencí. Pokud nemáme dostatečně bohatý vzorek, můžeme sekvence srovnávat s katalogem sekvencí z projektu 1000 Genomů nebo s jiným externím katalogem jako např. dbSNP 129 (databáze dbSNP 129 je často považována za poslední "čistý" dbSNP katalog bez mnohých vzácných a nepotvrzených variant z jiných velkoplošných projektů). Dalším krokem je hledání regionů, kde zatížení vzácnými variantami silně koreluje se zaměřením studie. Vzácné sekvenční varianty mohou mít různé charaktery - neutrální, protektivní a destruktivní (mutace), takže připadá v úvahu analýza variant z hlediska jejich přítomnosti v ne/kódujících oblastech, z hlediska jejich charakteru (synonymní a nesynonymní), z hlediska vlivu na fungování proteinů (predikce funkce proteinu -programy jako SIFT a PolyPhen2) apod.
NGS Lab, ngs@seqme.eu