
© SEQme s.r.o., 2012 - 2024. Všechna práva vyhrazena.
Právní upozornění.
webdesign Beneš & Michl
Short-read technologie je na maximu možného
Sekvenační technologie Illumina bývá běžně označována jako short-read technologie. V praxi to znamená, že typicky produkuje ready o délce řádově stovek bází. To je samozřejmě relativně málo a rozhodně to nenaplňuje potřeby, které vnímáme i mezi našimi zákazníky. Proto, jakkoliv Illumina stále vládne trhu a jistě ještě vládnout bude, máme v naší nabídce i alternativní technologie sekvenování, které mají délky čtení o několik řádů vyšší, takže tímto nedostatkem netrpí (a trpí třeba jinými).
Je celkem jasné, že pokud v budoucnu nějaká jiná technologie vytlačí Illuminu z pozice lídra trhu, bude to velmi pravděpodobně mimo jiné i kvůli délce čtení. V roce 2018 se společnost Illumina pokusila tento technologický problém „vyřešit“ převzetím jiné silné firmy v oblasti DNA sekvenování, společnosti Pacific Biosciences, která umožňuje na svých sekvenátorech čtení významně delší (více se o přednostech jednotlivých technologií můžete dozvědět na našich kurzech). Ale jak jste možná zaznamenali, na počátku roku 2020 bylo oznámeno, že obě firmy od tohoto záměru ustupují, převážně kvůli antimonopolním úřadům.
2x300 b na MiSeq
Je tedy celkem zřejmé, že chcete-li sekvenovat na Illumině, budete si muset i nadále vystačit s jejím aktuálním technologickým maximem co se délky čtení týká, a to je souprava pro přístroj MiSeq, která umožňuje čtení až 600 b (nastavení 2x300). Níže se stručně zabýváme souhrnem našich zkušeností se sekvenováním v tomto populárním nastavení, abyste vy, naši klienti, věděli, co můžete v takovémto případě čekat. (Mimochodem, z pokusu o akvizici Pacific Biosciences je celkem zřejmé, že Illumina delší ready zkrátka v dohledné době svým klientům nenabídne.)
Toto sekvenační nastavení samozřejmě funguje, ale důležité je říci, že pouze s určitým typem vzorků. Hlavní problém spočívá v tom, že výstupy jsou dle specifikace Illuminy počítány pro optimální, sekvenčně diverzifikovanou (typicky shotgunovou) knihovnu, což je využití, s kterým se v praxi setkáme málokdy. Typický klient pro sekvenaci tohoto typu je spíše zákazník v oboru metagenomiky, který chce sekvenovat amplikony. Jejich sekvenční diverzita je ale pochopitelně mnohem nižší než u knihovny získané náhodným štěpením nějaké genomické DNA. Vede to k tomu, že ke splnění technických specifikací máme zejména při sekvenování amplikonů velmi často daleko a není to v důsledku nějaké chyby (detailní rozbor níže). Proto jsme v roce 2019 tuto variantu sekvenace úplně vyřadili z nabídky, poněvadž jsme s naprostou většinou zakázek na 2x300 nebyli schopni splnit specifikace výrobce. Jenže následně celkem pochopitelně došlo k tomu, že jste se vy, naši klienti, neustále zajímali o možnost sekvenovat 2x300, protože to pro ty metagenomické knihovny prostě dává smysl, i když je kvalita či množství dat nižší než slibované Illuminou. Proto jsme se rozhodli v roce 2020 toto nastavení do nabídky vrátit, ovšem bez záruky co se týká množství dat i jejich kvality. Níže uvádíme, jaké výsledky zhruba můžete očekávat při sekvenaci v nastavení 2x300.
Specifikace výrobce k dnešnímu datu (3. března 2020 pro MiSeq 2x300):

Proč jsou amplikony problém?
V zásadě se dá říci, že jsou-li templátem amplikony, splnit uvedené specifikace lze pouze jednotlivě. Získat v jednom sekvenační běhu 44-50 milionů readů s délkou 300 b, kde by byla kvalita >70% bází vyšší než 30 (phred score) je dle našich zkušeností v podstatě nemožné. Pokud chceme co možná nejvyšší kvalitu, musíme snížit hustotu klastrů na sekvenační flowcelle, takže se to promítne v množství dat, která jsme vám schopni dodat. Pokud byste trvali na tom, že potřebujete těch zhruba 14 Gb dat, jak uvádí výrobce, bude to samozřejmě možné, ale v případě amplikonové knihovny nelze rozhodně počítat s kvalitou čtení dle specifikací. Hustota klastrů totiž bude muset být tak velká, že dojde k jejich vzájemnému „překryvu“, a kvalita prostě půjde dolů. Nemluvě o tom, že abychom „rozbili“ nízkou sekvenční diverzitu, musíme amplikony „naředit“ nějakou sekvencí s vysokou diverzitou (typicky PhiX), jejíž přítomnost pak bude významně snižovat objem dat, která vás zajímají.
A co s tím můžeme dělat
Svým způsobem jsme tedy částečně schopni ovlivnit sekvenační výstup nastavením sekvenačního běhu nebo charakterem sekvenační knihovny tak, abychom upřednostnili množství dat před kvalitou a naopak. V souhrnu jde tedy na naší straně o sladění následujících parametrů:
Uvedené parametry mají každý svou danou optimální hodnotu a při dodržení optimálních hodnot u všech parametrů můžeme vždycky teoreticky doufat v optimální výsledek. Nicméně dle našich zkušeností bude kvalita čtení posledních několika desítek bází z každého readu více či méně klesat. Pokud charakter experimentu neumožňuje optimální hodnotu některého z parametrů, je odklon od optimálního výsledku sekvenace obvykle spíše výrazný, což je typicky případ právě amplikonových knihoven.
Co můžete očekávat
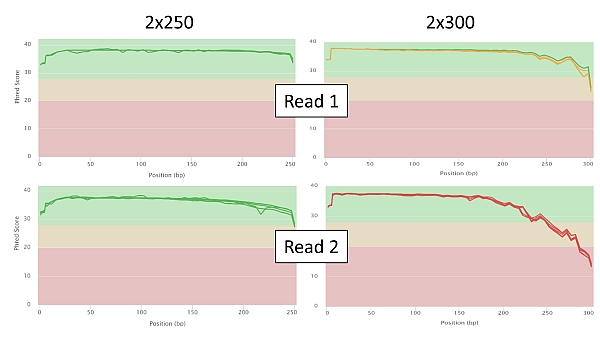
Níže uvádíme několik vybraných grafů kvality sekvenování v nastavení 2x250 a 2x300 pro srovnání. Z grafů závislosti Phred score na pozici té které báze readu je zcela zřejmý signifikantní pokles kvality čtení, jakmile se sekvenace blíží 250. bázi (PE250), nebo ji dokonce přesáhne (PE300). U readu 2 je tento efekt přirozeně výraznější.
Souhrn
Délka čtení 2x300 bazí je prostě pro tuto technologii v kombinaci zejména s amplikonovými knihovnami zatím nad její možnosti (a zřejmě to tak i zůstane) a na výsledky se nelze stoprocentně spolehnout, což je nicméně v případě technologie Illumina jediná zaznamenáníhodná výjimka.
V praxi to znamená, že pro sekvenování v nastavení 2x300 b, negarantujeme množství ani kvalitu dat, a to bez ohledu na typ knihovny. Na základě našich zkušeností jsme však schopni poskytnout vám kvalifikovaný odhad.
NGS Lab, ngs@seqme.eu