
 Analýza metagenomu
Analýza metagenomu
 Paralelní sekvenování amplikonů
Paralelní sekvenování amplikonů
 Celoexomové sekvenování
Celoexomové sekvenování
 Sekvenování technologií Illumina
Sekvenování technologií Illumina
 Long-read sekvenační technologie
Long-read sekvenační technologie
 NGS Services à la carte
NGS Services à la carte
 Sekvenátory v naší flotile
Sekvenátory v naší flotile
© SEQme s.r.o., 2012 - 2024. Všechna práva vyhrazena.
Právní upozornění.
webdesign Beneš & Michl

Vy dodáte RNA, knihovny nebo data. My dodáme výsledky analýzy diferenciální exprese v publikační kvalitě.
Analýza diferenciální genové exprese, běžně zkracovaná jako DG nebo DGE analýza, je analýza rozdílů v četnosti výskytu genových transkriptů v transkriptomu podle fenotypu nebo experimentálních podmínek. Cílem analýzy diferenciální exprese je určit, které geny jsou odlišně exprimovány mezi porovnávanými podmínkami. Tyto geny mohou nabídnout biologický náhled na procesy ovlivněné podmínkou (podmínkami) zájmu.
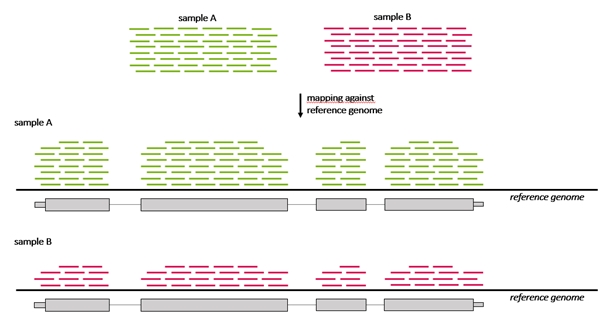
Protože počet genů, které jsou rozdílně exprimovány mezi vzorky, může být vysoký, je nutná metoda k pochopení a interpretaci významu tolika změn genové exprese. Metoda musí umožnit seskupení genů, které patří do konkrétní kategorie a mají odlišnou hladinu exprese v jednom vzorku ve srovnání s jiným vzorkem. Například pokud má vzorek rakoviny prsu odlišně než kontrolní vzorek exprimováno více genů, které jsou všechny anotovány jako „geny buněčného cyklu“. Seskupení genů lze provést na základě řady zdrojů, jedním z nich je Gene ontology databáze (GO).
Gene Ontology (GO) databáze je největším zdrojem informací o funkcích genů na světě. Tyto informace jsou dostupné/čitelné člověkem i strojově a tvoří základ pro výpočetní analýzu rozsáhlých molekulárně biologických a genetických experimentů v biomedicínském výzkumu. GO definuje pojmy / třídy používané k popisu funkce genů a vztahy mezi těmito pojmy.
Analýza DGE v naší laboratoři proto zahrnuje:
Pokud již máte nezpracovaná data od jiného poskytovatele sekvenačních služeb a potřebujete pouze pomoci s jejich analýzou, jsme vám také k dispozici.
Poznámka: Seznamy genů, které se liší mezi dvěma nebo více sadami vzorků, jsou často získány technologií RNA-seq. RNA-Seq umožňuje sledovat nejen změny v genové expresi v průběhu času nebo rozdíly v genové expresi v různých skupinách nebo léčbě. Tuto techniku můžete použít také ke zkoumání alternativního sestřihu, post-transkripčních modifikací, genových fúzí nebo mutací / SNP. Pokud jsou toto vaše cíle a potřebujete analýzu vzorků nebo dat, neváhejte nás kontaktovat.
Množství dat potřebných na vzorek lze určit konceptem hloubky. Například vzhledem k tomu, že lidský transkriptom tvoří 3% lidského genomu (3 Gb), znamená 90 Mb dat hloubku 1× a tedy průměrné pokrytí každého exprimovaného nukleotidu jednou. Některé geny jsou však exprimovány mnohem silněji než jiné a některé geny naopak málo, takže i hloubka 1000× by poskytla jen rovnoměrnou šanci na sekvenování transkriptu, který je v buňce 1 na tisíc.
Množství potřebných dat proto závisí na strategii přípravy knihovny (ribodeplece, polyA selekce, …), zdrojovém organismu a velikosti jeho transkriptomu / genomu a genech, na které se chceme zaměřit (ať už předpokládáme vysoké nebo nízké úrovně jejich transkriptů) ).
Pro DGE lidských vzorků se obecně doporučuje nejméně 30 mil. readů na vzorek a v případě nemodelového organismu může být zapotřebí provedení pilotní studie.
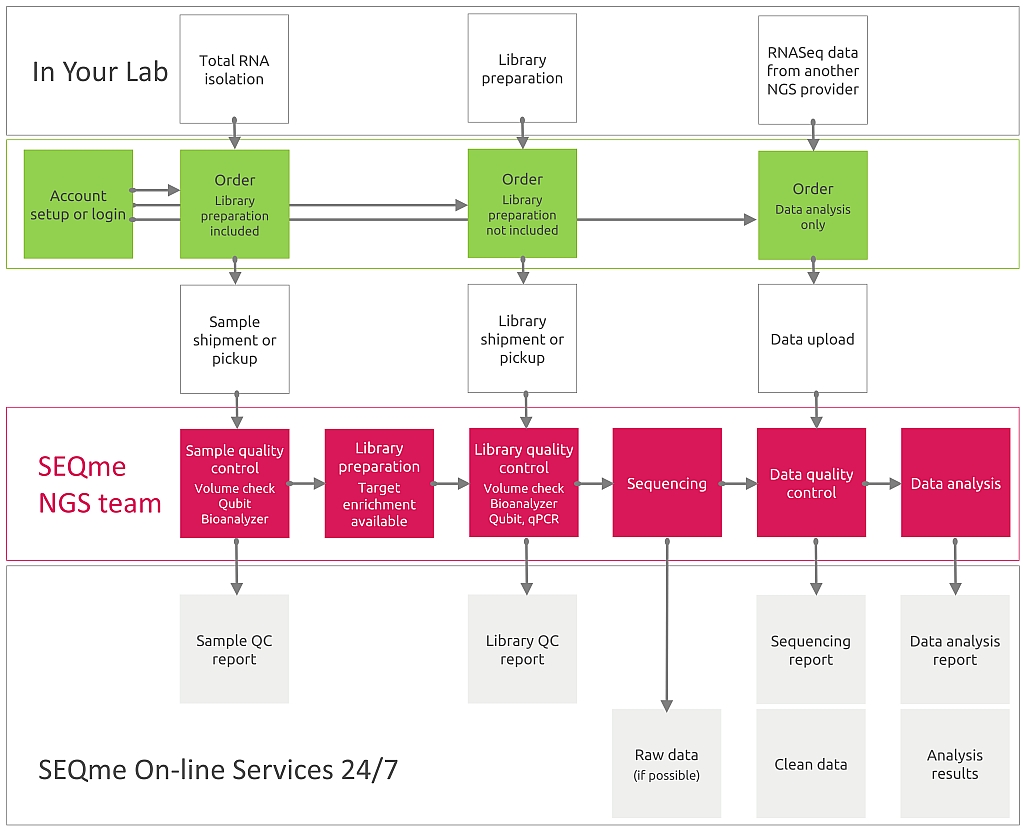
Požadujete-li laboratorní zpracování, dodáváte RNA nebo hotové sekvenační knihovny. Chcete-li objednat pouze analýzu dat, čtěte níže.
Musíte provést izolaci RNA. V naší laboratoři budou vaše vzorky zpracovány následně takto:
Získané sekvence budou roztříděny dle kombinace indexů do souborů reprezentujících jednotlivé vzorky a bude provedena analýza ukazatelů kvality sekvenace, jako je počet a délka sekvencí, phred score, %GC, úroveň duplikace atp.
Jako výstup obdržíte data ve formátu FASTQ rozdělená do souborů dle jednotlivých vzorků.
Požadavky na provedení analýzy dat:
Je důležité si uvědomit, že gen je považován za diferenciálně exprimovaný, pokud je pozorovaný rozdíl hladiny jeho exprese mezi dvěma experimentálními podmínkami statisticky významný, tzn. pokud je rozdíl větší než to, co by se dalo očekávat jen kvůli náhodným změnám. DGE je tedy statistická technika a jako taková musí splňovat základní statistické požadavky týkající se počtu vzorků / skupin k porovnání. K úspěšnému provedení DGE na vašich datech je proto nezbytné:
Výstupy (eng):
Dodržujte naše Pokyny pro přípravu vzorků. Pro úspěšné provedení analýzy dat je zapotřebí alespoň 6 vzorků / datových souborů.
Upozorňujeme, že úspěch analýzy velmi závisí na integritě RNA, kterou nám dodáte! Použití degradované RNA může vést k problémům při přípravě standardních sekvenačních knihoven. Pokud nemůžete izolovat RNA o vysoké integritě (RIN>7), zvažte objednání knihoven typu 3' mRNA library (QuantSeq/UMI), kde nejsou požadavky na kvalitu materiálu vysoké a je tedy možná i analýza degradované RNA.
Analýzu lze objednat včetně laboratorního zpracování vzorků nebo pouze jako analýzu dat. Chce-li objednat laboratorní zpracování technologií Illumina, volte možnost Illumina - Sekvenování na zakázku.
Chcete-li objednat pouze analýzu dat, volte Bioinformatické služby.