
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Just as few people go to the theater or a sports match without knowing what is on the program, it is normal for any other service you intend to order to have at least some idea in advance of what you can expect from the service provider, especially in terms of QUALITY, SPEED and PRICE. This of course also applies to DNA sequencing. It is understandable that most clients would like all of these three criteria be met at 100% at the same time, but we do say that this is not basically possible. If anyone claims to be able to sequence your samples at the lowest price on the market in the shortest possible time while at the same time achieving top quality, they are not honest with you. This is depicted by a (generally known) diagram:
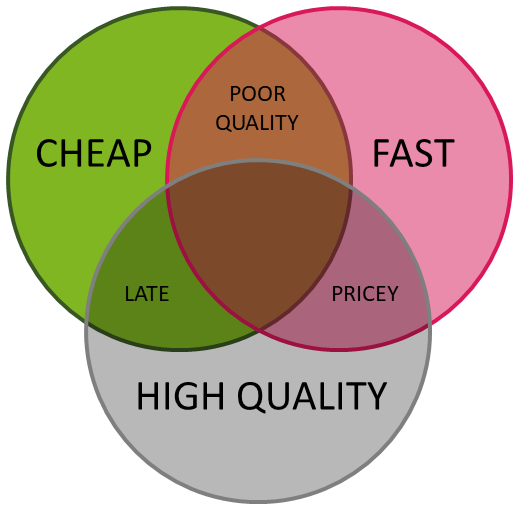
Sequencing service providers are betting on different approaches, but quite often they tend to be 'cheap and fast'. However, we dare to say that for sequencing analyzes not always the faster is the better! The reason is simple - sequencing tools (sophisticated instruments, software equipment, etc.) are great helpers, but to achieve the highest quality of sequencing, more work is needed that only qualified people can do and this requires a certain amount of time. We talk in particular about multiple checks (receipt and control of samples, workflow, resulting data, etc.), operational solutions for possible problems, common communication and customer consultation, or taking into account the specific requirements for sequencing analyzes (laboratory as well as data requirements). We know from experience that only with this approach can we deliver high quality results, while ultimately saving our customers time and money, and it is a great challenge for us to get closer to this ideal.
Different approaches to sequencing data analysis
In the context of Sanger sequencing, especially the data analysis pipeline may be a good example of a variety of providers’ approaches.
Sequencing data (electropherograms and sequences) can be the result obtained either solely by automated analysis (without human eye control or human hand intervention) or including this control / intervention by an experienced operator (which is the SEQme way of doing things).
To explain this, we need to get a little bit into the technical details of the process. In simple terms, automatic sequencing analysis is performed using the Data Collection software of a particular sequencer and then typically (but not exclusively) Sequencing Analysis software, which includes so-called basecallers (algorithms) for sample analysis. This process includes setting all parameters necessary for raw data analysis and sequence "generation" (i.e. basecalling), such as setting the start and end points of analysis, spacing, mixed-base detection settings, and many others. Generally, it can be said that the auto-analysis of most samples have good results. However, there are quite a number of cases where, by manual editing and reanalyzing (including the use of paid software tools), we obtain results in significantly higher quality than automatic analysis is capable to provide!
Therefore, the evaluation of the quality of your sequences in SEQme is always first performed by machines and the automated software, but the data thus evaluated are always and without exception additionally evaluated by humans. Manual (human) analysis of sequencing data naturally requires know-how, experience and time, but because we believe that providing the sequencing service is primarily about the quality, credibility and maximum usability of the resulting data and only then about the speed of delivery, since the day we started to provide our services we go this way.
UPDATE: As of October 2022, we will be adding the RapidSeq protocol to our Sanger portfolio, which is slightly inconsistent with the philosophy described above. The evaluation of results obtained by the RapidSeq protocol takes place without the intervention of human hands! Each result is algorithmically processed immediately after completion in the sequencer and automatically sent to clients. By including this protocol, we are trying to cope with demands of our clients for whom the time to results is a critical parameter!
What, then, does the manual / visual evaluation of sequencing data mean, what do we get and in what cases?
Below are some examples of which the benefit is clearly apparent. All images always show the result of automatic analysis first and then the result obtained from the same sample after operator intervention!
• PeakTrace - reliable data above 850 bp?
PeakTrace is a paid basecaller that recalculates your .ab1 files using its own set of algorithms, which in some cases effectively improves the display of individual bases and extends the read length. We know this software and its tools very well and based on our experience we can decide when its use is beneficial because it is definitely not always. If we believe it makes sense, we provide free PeakTrace analysis.
Since the capillary electrophoresis of the automated DNA sequencers is optimized for reading fragments approximately 850 bp long, the reliability and usability of the sequencing data is gradually deteriorating beyond this area (broad peak shape, overlapping peaks and especially low quality score). While the automatic basecaller is inaccurate or even error-prone in these cases, PeakTrace achieves high-quality reading up to approximately 1200 bp. It can therefore be used in particular for longer PCR fragments or plasmids. PeakTrace can also accurately identify and quantify polymorphic sites in the sequence, or remove undesirable data (sequencing electrophoresis artifacts, background noise caused by the minor presence of by-products in the reaction, etc.).
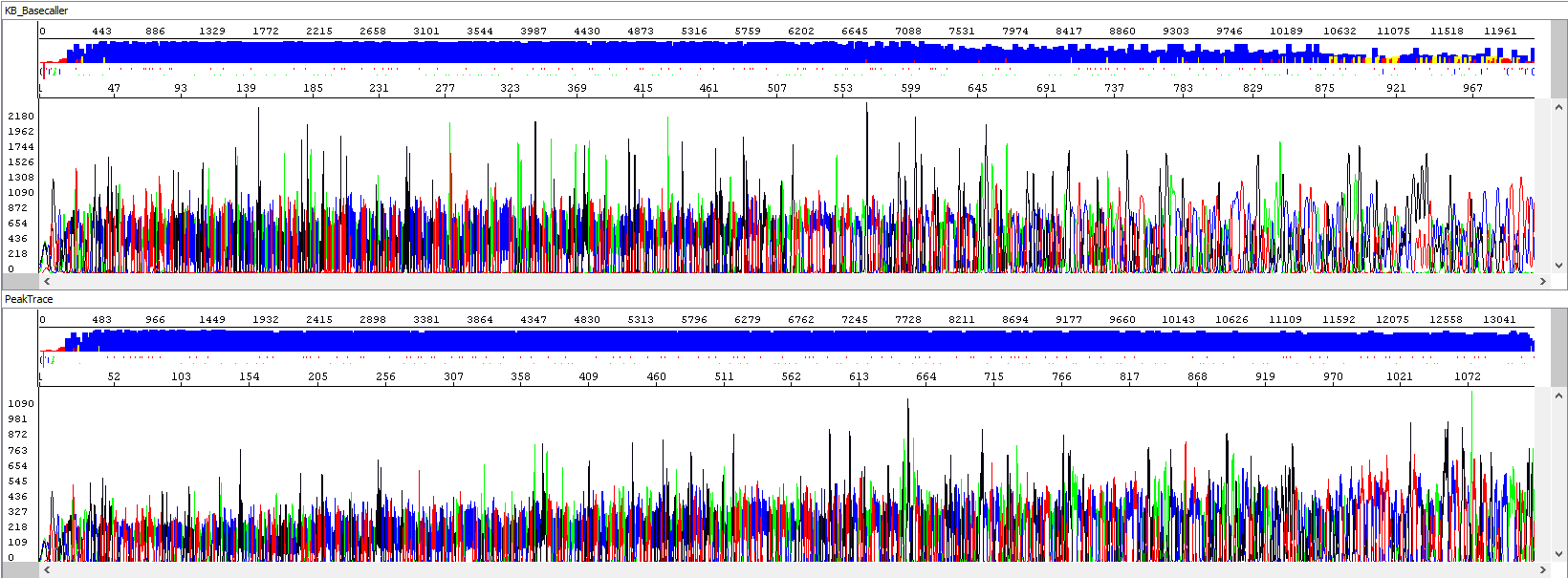
Example 1 - Optimization of read length, upper window showing standard algorithm analysis (read length 934 bp, QV20), bottom window the same sample after PeakTrace algorithm reanalysis (read length 1113 bases, QV20, 19% longer sequence):
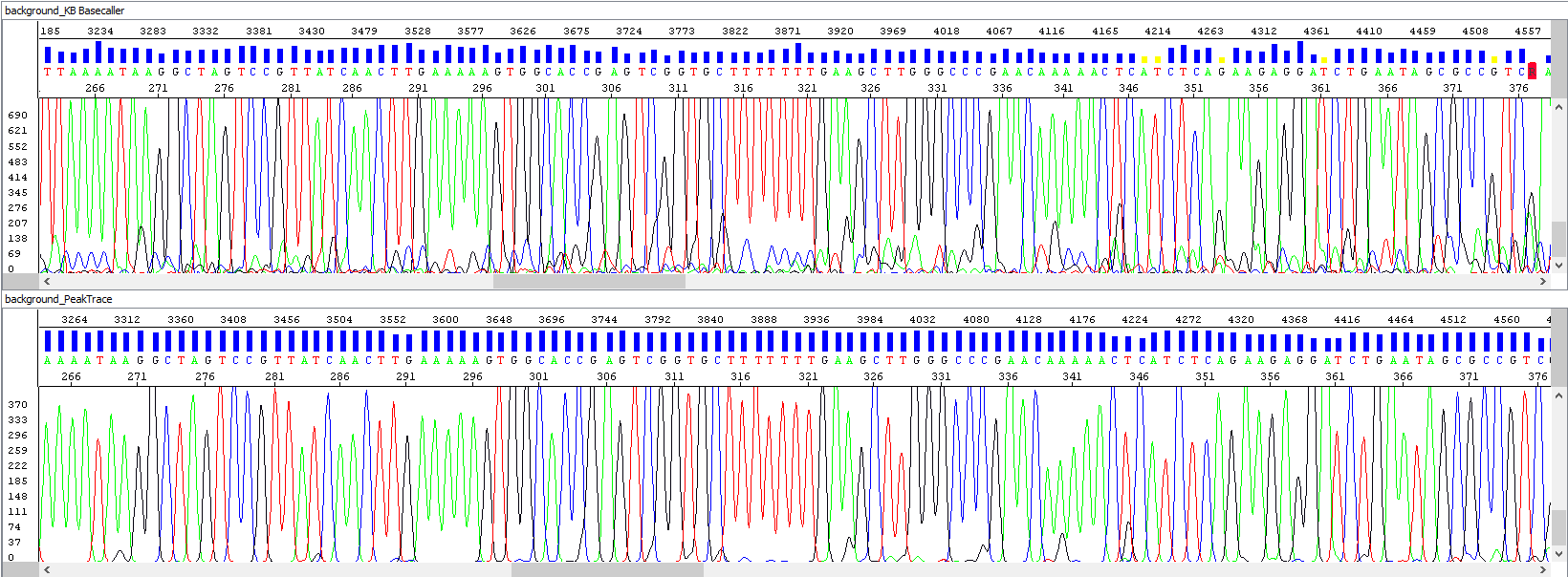
Example 2 - Background signal optimization, upper window showing standard algorithm analysis, bottom window the same sample after PeakTrace algorithm reanalysis, note the background of sequences, the picture speaks for itself:
• Short read length - actual end of sequence?
Some sequencing reactions are terminated abruptly and prematurely. Possible reason may be the presence of the secondary structure (hairpin) or GC rich area in the analyzed sequence (more about this issue here). Often, the sequence in the problematic section actually ends (the polymerase does not read), but there are cases where the fluorescence signal drops significantly, but the sequence continues (the hairpin is at least partly read). Without a subjective scan of raw data and manual editing of analysis parameters, the usual result of automatic analysis is only a short sequence ending with the problem area. It would be a pity to lose the data beyond this area, so we always re-analyze these sequences manually to get as much reading as possible.
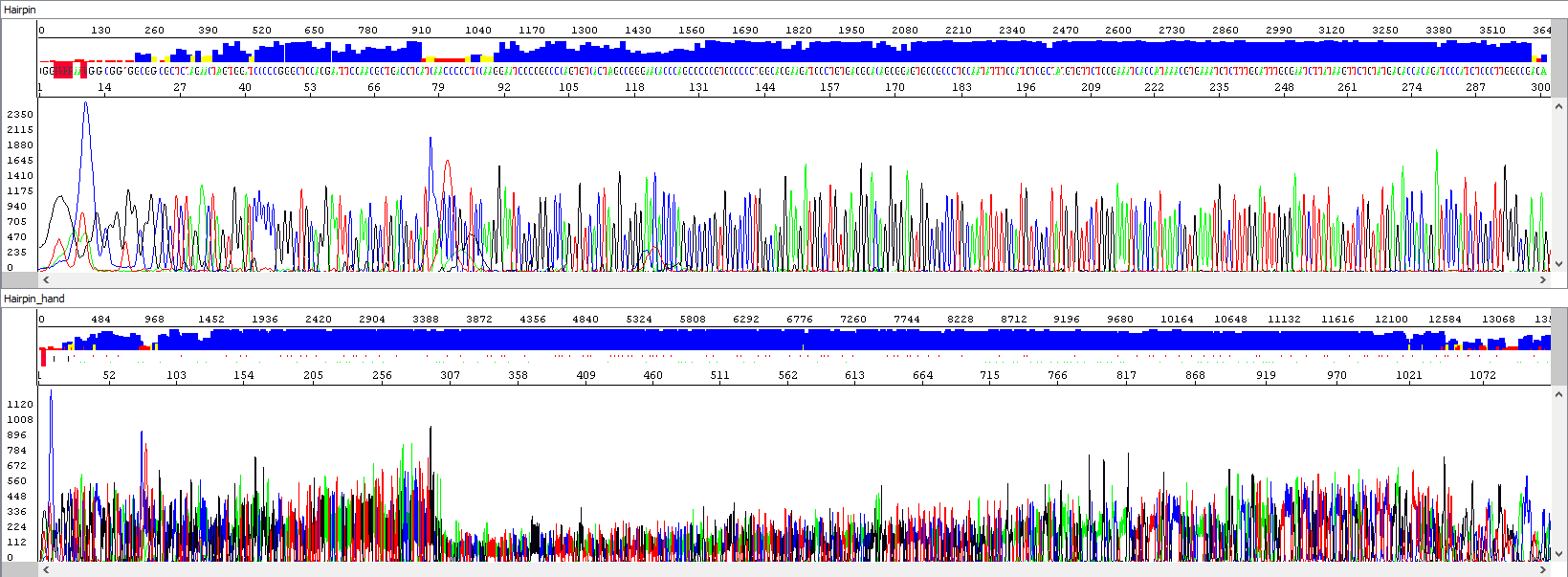
Example 3 - Optimization of the read length in the presence of a hairpin; upper window displaying a standard analysis by an automatic algorithm (read length 284 bases, QV20), bottom window after manual intervention by the operator, who extended the read length and simultaneously used PeakTrace algorithm reanalysis, read length 1106 bases, QV20, 389% longer sequence):
Showing also raw data - identical in both windows:
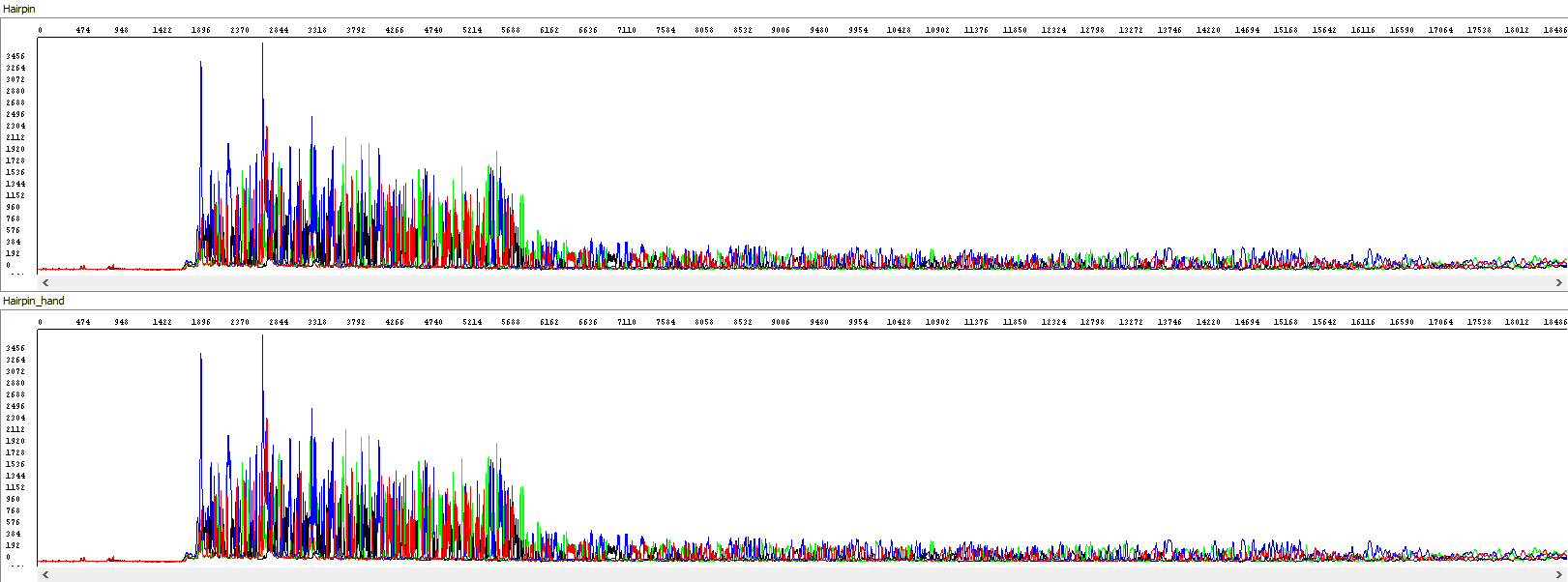
From the thumbnail, it is clear that the operator only utilized the sequence that the automatic algorithm simply did not use.
• Primer dimers - Unreadable Beginning
In some cases, we encounter very short unwanted fragments at the beginning of the sequence. These may be primer dimers or other non-specific products of PCR or sequencing reactions. Since they are usually in surplus, there is a depletion of the labeled nucleotides at the beginning of the sequencing reaction to produce these short products, and for the desired sequence nothing is "left behind". The result is a sequence with an unreadable beginning of high signal strength (where the individual peaks overlap each other), and poor visualization and the quality of the desired sequence. We manually modify such data, especially by removing the mixed sequence at the beginning.
Example 4 - Optimization of the read length in the presence of primer dimers, the upper window displays a standard analysis by an automatic algorithm that is totally unusable (read length 0), the bottom window after re-analyzing by an experienced operator (read length 863 bases, QV20):

Showing also raw data, zoomed in - there is a huge primer peak, identical in both windows and only then the correct data can be read (relatively weak, but at least something, we read out 863 bases, but only when we step into the process manually):
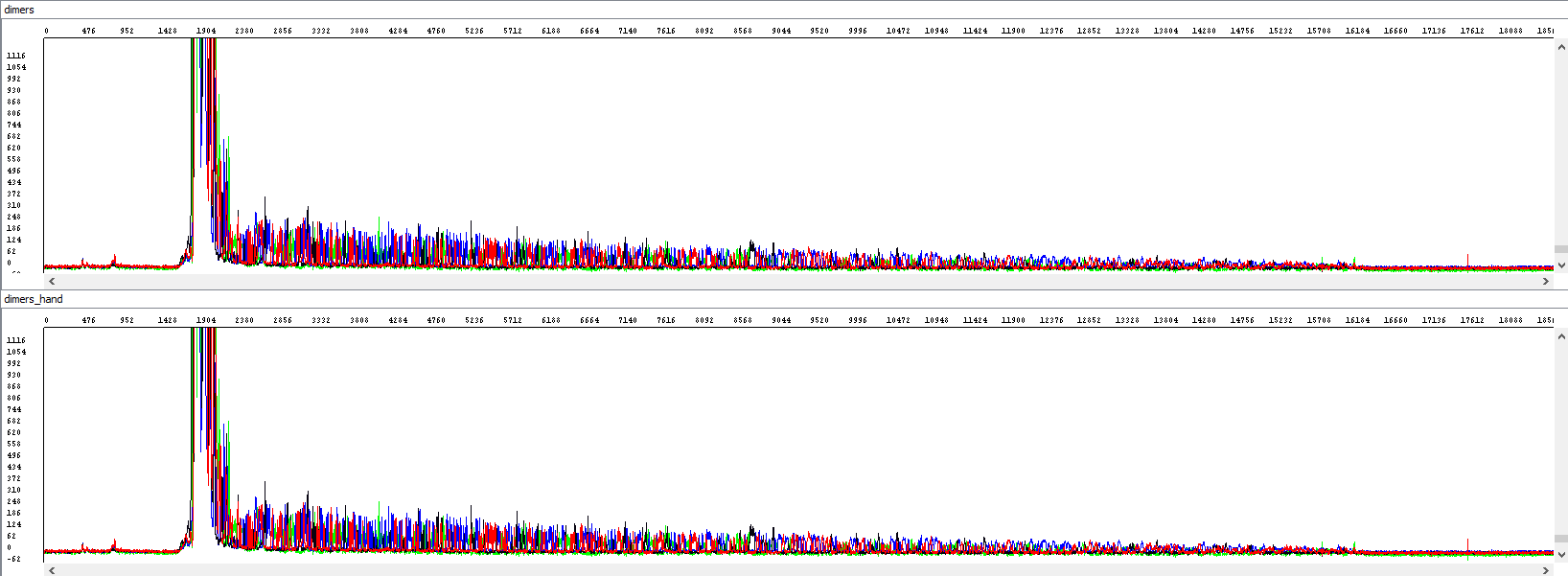
• Late sample migration in the capillary and chances for a good sequence
For some sequences, the signal begins later than expected, and there is a rapid loss of resolution of individual bases (observable especially in raw data). The cause may be suboptimal injection of the sample into the capillary (in fact a technical error of the machine solvable by repeating the analysis), but most often it is the presence of non-specific inhibitors in the sample that interfere with capillary electrophoresis and cause this delayed migration. This, of course, reflects on the quality of the data, in which the individual bases cannot be reliably distinguished (the result is similar to the loss of resolution demonstrated below, Example 5). The automatic basecaller does not know this naturally, but we do, and in these cases we usually get the perfect sequence after proper sample dilution and repeated electrophoresis. In fact, of course, it means later delivery of the result, because the analysis simply needs to be repeated.
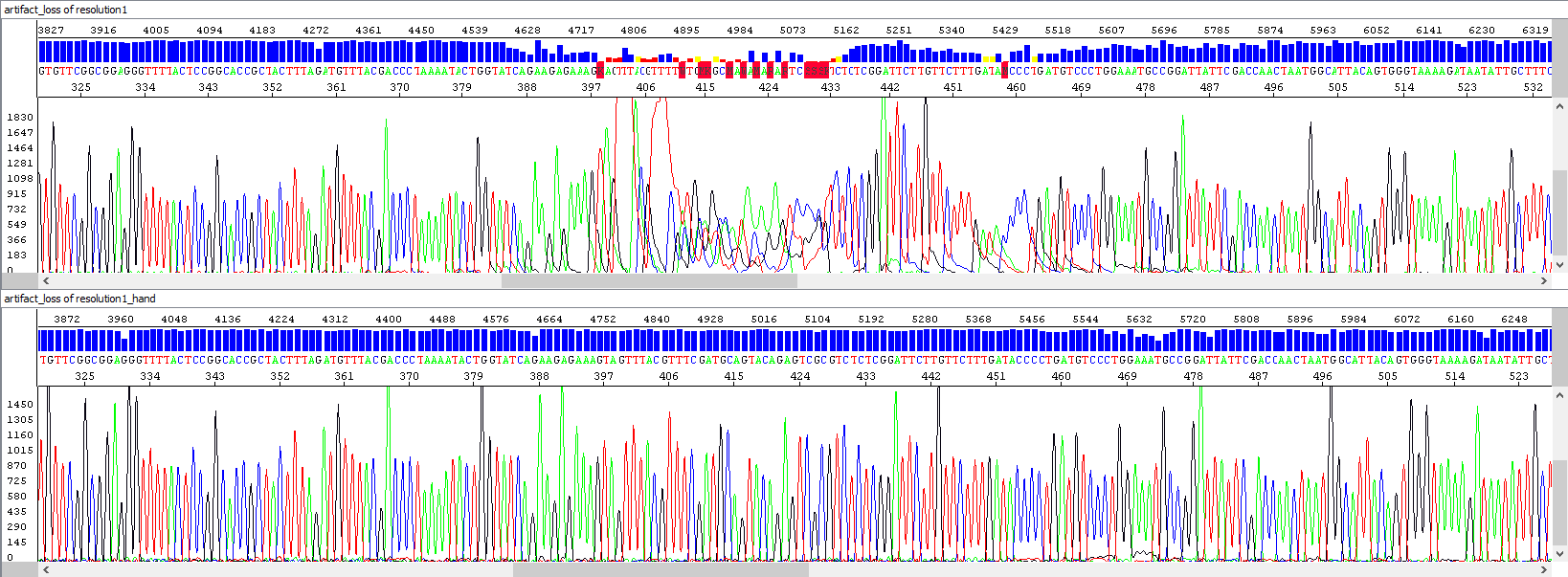
• Sequencing artifacts
Automatic sequencers are very precise and sensitive, but sometimes there are certain separation artifacts in the resulting data, which may negatively affect the interpretation of the analyzed sequence (in extreme cases the artifact may be considered as a nucleotide variation in the DNA sequence, mutation). We strive to minimize these phenomena - of course, by providing the best possible care to our sequencers and also by visually checking each sequence that passes through the capillary. If we encounter this problem, we automatically reiterate the analysis as quickly as possible.
The most common artifacts include, for example, the loss of resolution in the middle of the sequence, or the so-called spikes (multicolored overlapping peaks) that occur in the sequence in a random fashion. They are most often caused by small air bubbles, microcrystals inside the polymer in the capillary, or imperfect polymer exchange between runs. There may, of course, be more to the causes, but there is certainly a link between the quality of the template and the extent to which the eventual separation artifacts may affect the resulting sequence - in weak signal samples (e.g. with low template concentration), these problems will manifest much more significantly.
Example 5 - Repeated analysis (bottom window) after loss of resolution (upper window), probably due to contamination:
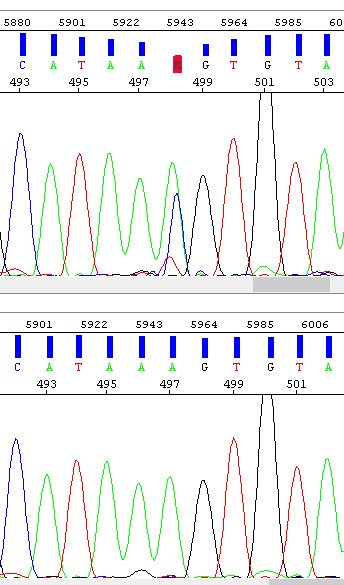
Example 6 - Spike, the result of which is the "mutation" at position 498. The operator encountered this problem and repeated the analysis (bottom window) - the result is a clean sequence.
Unsuccessful sequencing - we do really care
Unfortunately, not all sequencing analyzes come out optimally or even worse - they do not come out at all. Although we cannot know the background of all of the sequencing analyzes we perform (in terms of the goal of the experiment and the area of interest), we can often identify the problem based on the analyzed data. Since we look at your data very carefully, it is not unusual to get our observations as a comment when sending results to you (these are most often related to concentration of templates, mixed templates, probable presence of sequencing inhibitors, primer dimers, homopolymers, repeats, secondary structures, or GC rich areas of the template, etc.). We are glad to help you with this advice to get better results.
In summary, it is true for all of the examples above that when evaluated by automatic algorithms sometimes you will get good results but sometimes not. Experienced operator is always evaluating them correctly. We at SEQme believe that this is exactly the reason why our experienced operators must look at your data! In conclusion, we would like to say that the issues described above are solved automatically in our labs sometimes without even informing you, so if you feel that you have never seen the problematic data, that's precisely because :-)
Sanger lab, info@seqme.eu