
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Assuming we have good signals and read length is as expected which means we have successfully solved all issues mentioned in previous two parts of this post, we can still be far away from seeing nice data. This third part deals with the problem of having peaks overlapping other peaks. The sequence is not readable.There are in principle three possible outputs having varying causes:
More than one priming site
If your template is long enough and your primer binds at two sites, both far from its end, you will obtain a mixed sequence from the very beginning to the very end. If, on the other hand, the sequencing primer anneals to a second site near the end of PCR product you can get a mixed sequence only at the beginning and then, once one of the sequencing reactions reaches the end of the template, only a clean sequence proceeds onwards.
Mixed templates and/or primers
Two templates plus one primer or two primers plus one template or, at the worst, several templates and several primers. If this happens after sequencing PCR products, find out what is going on and proceed accordingly. The error is probably yours, typically unsatisfactory template cleanup. If this happens after sequencing plasmid DNA, be aware that plasmids that most people use these days have very high copy numbers. Taken together with high efficiency of transformation of E. coli, even if you grow a single colony after transformation it can still produce DNA that is a mixture of different plasmid molecules. We recommend to pick a single colony after transformation and streak it out at least once for single colonies.
Double peaks with special pattern (n+1 or n-1)
Contamination of the sequencing primer with n+1 or n-1 primer. Your primer is in fact a mixture of two – the desired primer and another one which is one base longer or shorter. You should ask for a resynthesis of this primer. This is not a mistake of yours, this is a mistake of the company which provided the primer. Shame on them!
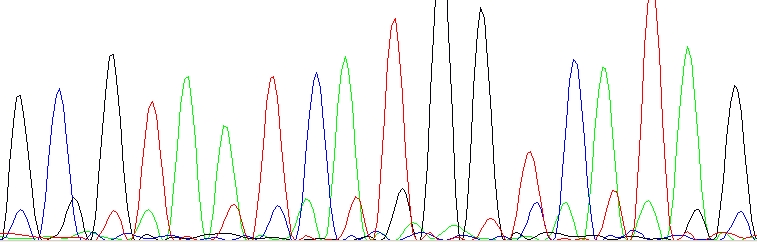
n+1 or n-1 primer – The lower sequence is same as the upper sequence, only shifted one base left
Primer dimers
The sequencing or PCR primer(s) formed a dimer during the PCR and/or the DNA sequencing reaction and produced a short product that is sequenced at the same time as the normal target. This may be resolved by redesigning the primer or choosing a proper cleanup strategy (see above).
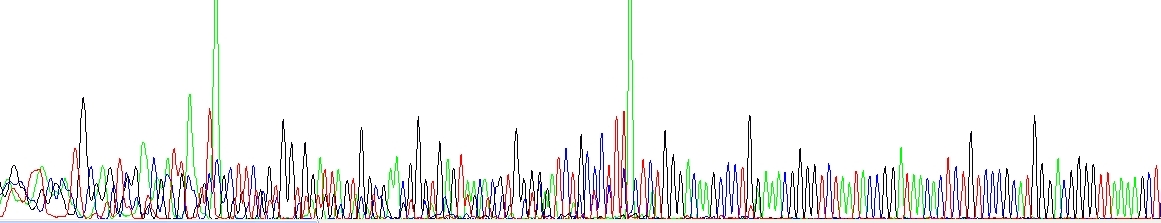
Primer dimers and/or other short unspecific products. Please notice very strong green peaks at the end of each unspecific product, this is a consequence of the Taq enzyme adding A at the 3'-end and it is quite symptomatic.
More than one priming site – See above
Mixed templates and/or primers – See also above
After a homopolymer or repeats
This is a rather peculiar case, the electropherogram is very characteristic. Once I heard someone to call this “running hedgehogs” :-D which seems very appropriate.
Taq DNA polymerase used for PCR and/or DNA sequencing is not very processive. This means that after polymerizing a short stretch of DNA (about 35 nucleotides on average) the enzyme dissociates from the template, taking the 3’ end off of the template. (In contrast, T7 DNA polymerase can extend about 1,000 nucleotides and the replicative DNA polymerases millions of bases before dissociating from the template. Unfortunately, only Taq DNA polymerase can be used for cycle sequencing.) If this happens in the homopolymer region and the 3’ end of the dissociated extension product re-anneals back, it may anneal at exactly the correct position or it may anneal shifted by a base forward or backwards from its correct alignment. Consequently, when the product extends out of a homopolymer region it may be one or more bases shorter or longer than its template sequence. When these extension products are electrophoresed on a DNA sequencer, the sequence up to the homopolymer region is clean, but after this region the sequence appears mixed. The severity of this mixed sequence will increase with the length of the homopolymer stretch.
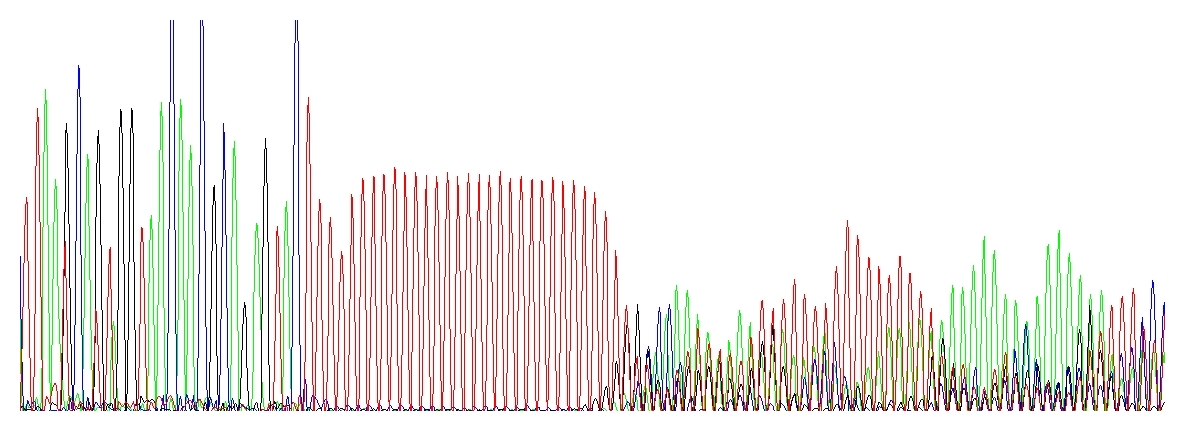
„Running hedgehogs“ catching up a red polyT stretch
It is usually very challenging to solve this problem to the investigator’s satisfaction. In a plasmid or a restriction fragment isolated from a plasmid, one can sequence across as many as 15 repeated nucleotides before mixing becomes significant. In a PCR product you will observe mixing occurring after only 8–10 repeated nucleotides and sometimes even less because the polymerase stutter occurred during both the PCR reaction and the sequencing reaction. You can therefore avoid PCR and try cloning the fragment into a plasmid and sequence the plasmid then. Alternatively you can design a primer that contains the homopolymer region. If the region is for instance a poly A, you can design oligo dT primer with either a C, A, or G as the 3’-terminal nucleotide. Similar strategy can be used for the reverse strand. These primers allow one to sequence out from the homopolymer and using flanking primers one can sequence into the homopolymer region. By this method, you should obtain most of the sequence. However, the actual number of bases in the homopolymer may still be difficult to determine.
After clean sequence (no homopolymer or repeats) – Insertion or deletion
Up to this point the sequence is clean, from this point onwards it is not. Your template is in fact a mixture of two templates and one of them contains an insertion or deletion (one base or longer). Sequencing using both forward and reverse primer is usually necessary.
Of note, the points I mentioned in these posts are also part of our DNA sequencing seminars. We organize these events upon request in your institute to bring all researchers to the same level and to standardize best practices. Their timeline and agenda range from a brief presentation focused on basics to very detailed trainings lasting days. Have a look at our training website.
Sanger lab, info@seqme.eu