
 Metagenome sequencing
Metagenome sequencing
 Tailed amplicon sequencing
Tailed amplicon sequencing
 Whole exome sequencing
Whole exome sequencing
 Illumina sequencing
Illumina sequencing
 Long-read sequencing technologies
Long-read sequencing technologies
 NGS Services à la carte
NGS Services à la carte
 Instruments in our fleet
Instruments in our fleet
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl

You submit either RNA, libraries or data. We provide publication-ready results of differential gene expression analysis.
Differential gene expression, commonly abbreviated as DG or DGE analysis refers to the analysis and interpretation of differences in abundance of gene transcripts within a transcriptome according to phenotype or experimental conditions. The goal of differential expression testing is to determine which genes are expressed at different levels between conditions. These genes can offer biological insight into the processes affected by the condition(s) of interest.
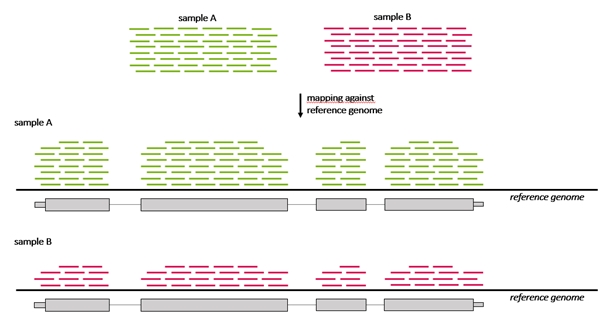
Because the count of genes that are differentially expressed between samples may be high, a method to understand and interpret the meaning of so many gene expression changes is needed that enables grouping of genes that belong to a particular category enriched in one sample compared to another sample. For example, if a breast cancer sample has more genes regulated that are annotated to the “cell cycle genes group” than a control sample. Grouping of genes can be performed based on their annotation to a number of sources, one of these being The Gene ontology resource (GO).
The Gene Ontology (GO) resource is the world’s largest source of information on the functions of genes. This knowledge is both human-readable and machine-readable, and is a foundation for computational analysis of large-scale molecular biology and genetics experiments in biomedical research. The GO defines concepts/classes used to describe gene function, and relationships between these concepts.
Our DGE lab & data analysis pipeline therefore includes:
Should you have raw data from another provider already and need only help with its analysis, we are at your disposal too.
Note: Lists of genes that differ between two or more sample sets are often provided by RNA-seq data analysis tools. RNA-Seq enables to look not only at changes in gene expression over time, or differences in gene expression in different groups or treatments. You can employ this technique to investigate also alternative gene spliced transcripts, post-transcriptional modifications, gene fusion or mutations/SNPs. If these are your scientific goals and you need sample or data analysis, do not hesitate to contact us.
The amount of data needed per sample can be determined by the concept of depth. For example, given that the human transcriptome accounts for 3% of the human genome (3 Gb), having 90 Mb data would be 1×depth and on average cover each nucleotide of interest once. However, some genes are more highly expressed than others and some genes are rarely expressed, so even 1000×depth would only provide an even chance of sequencing a transcript that is 1 in a thousand in a cell.
The amount of data needed therefore depends on the library preparation strategy (ribodepletion, polyA selection, …), the source organism and the size of its transcriptome / genome and the genes we want to target (whether we assume high or low levels of their transcripts).
At least 30 mil. reads for DGE of human samples is generally recommended and a pilot study might be needed in case of non-model organism.
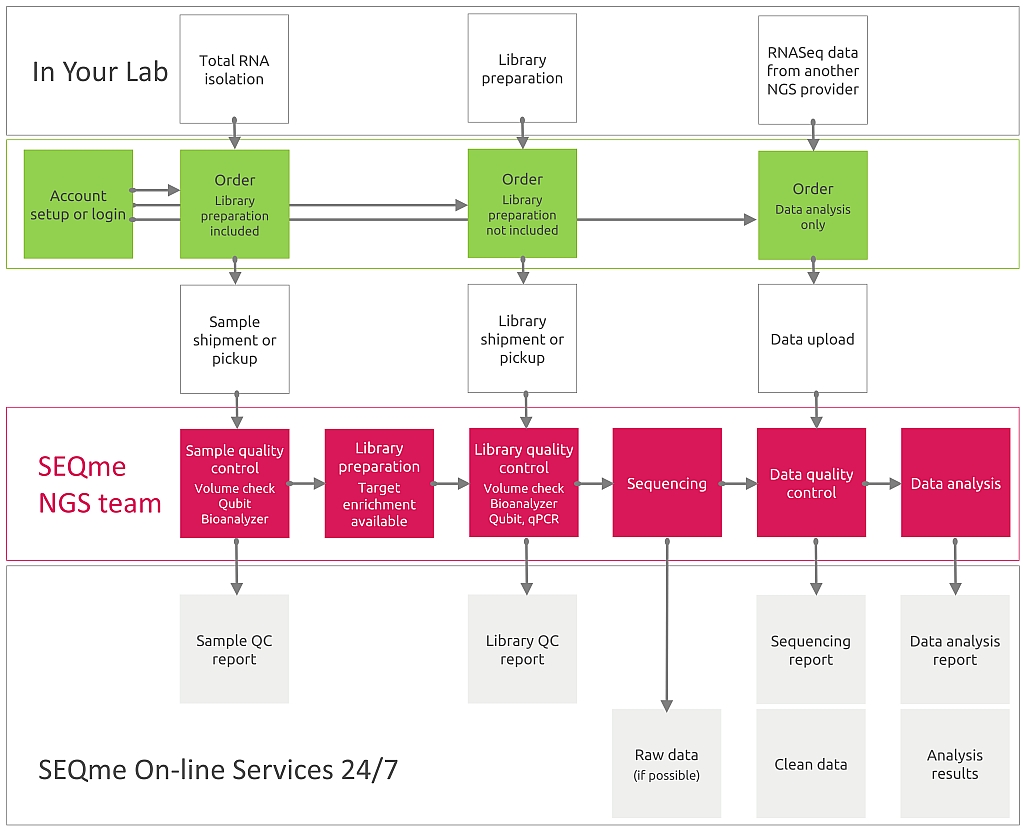
Should you request wetlab processing of your samples, you must provide RNA or sequencing libraries. If you need data analysis only, please scroll down.
For wetlab processing you must perform RNA isolation. Then, in our lab your samples will be processed as follows:
The sequences obtained will be sorted according to the combination of indexes into files representing individual samples and analysis of sequencing quality indicators such as count and length of reads, phred score, %GC, duplication level, etc. will be performed.
As output you will receive data in FASTQ format divided into files according to individual samples.
Requirements to perform data analysis:
It is important to understand that a gene is declared differentially expressed if an observed difference (change in read counts) between two experimental conditions is statistically significant, that is if the difference is greater than what would be expected just due to random variation. Therefore, DGE is a statistical technique and as such must meet basic statistical requirements regarding count of samples/groups to compare. To successfully perform DGE on your data, these are the “musts”:
Data analysis outputs:
Follow our Sample submission guidelines. To perform data analysis, at least 6 samples / data sets are required.
Please note that the success of the wetlab analysis depends very much on the integrity of RNA you provide! The use of degraded RNA can compromise standard library preparation. If you are not able to isolate high-RIN RNA (>7), you may think of ordering 3' mRNA library prep (QuantSeq/UMI) instead where the quality requirements are not strict and allow analysis of degraded RNA samples.
The analysis can be ordered and commissioned online including wet lab sample processing or data analysis only. If you intend to order sample processing by Illumina technology, select Illumina - À la carte sequencing.
If you intend to order data analysis only, select Data analysis services.