If you want to analyze the presence of microorganisms in a particular environment, metagenomic sequencing is a very effective first choice tool, as it allows identification of all species in test samples.
The most commonly used strategy is based on sequencing of amplicons obtained by amplifying selected genes or hypervariable regions of these genes. Typically these are ribosomal subunit genes (such as 16S, 18S or ITS) but not only. These targets are traditionally used to identify organisms by comparing the sequences obtained with reference databases.
An alternative approach is shotgun sequencing of the whole sample or metatranscriptomic analysis. If you are interested in these services, please contact us.
Metagenomic analysis using amplicon sequencing
| Resolution |
Genus level |
Species level |
| Technology |
Illumina |
Oxford Nanopore |
| Target |
Hypervariable region of a selected gene |
Selected gene |
| Data amount you will get |
From 20.000 reads/sample |
From 10.000 reads/sample |
| Time to results |
Approx. 4 weeks |
Approx. 10 working days |
Comparison of Illumina and Oxford Nanopore outputs: Illumina technology is capable to discriminate down to the Bifidobacterium genus level whereas by using Oxford Nanopore technology we can identify species, e.g. Bifidobacterium breve.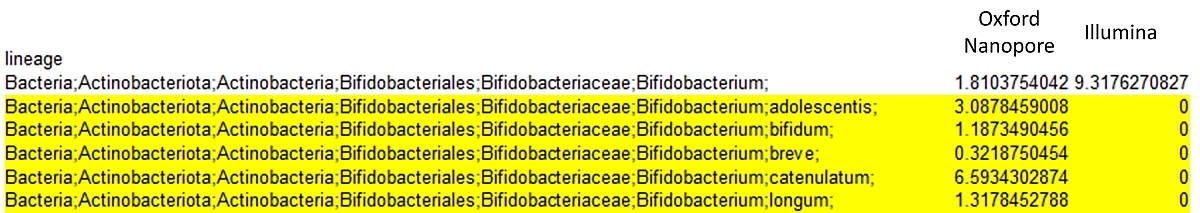
Laboratory processing
Our laboratory strategy is designed to be robust enough for different sample types. Your samples will be processed as follows:
- Amplification of selected genes or hypervariable regions
- Preparation of sequencing libraries
- Quantification of libraries to achieve maximum balanced sequencing yield
- Sequencing and data analysis (optional)
What primers do we use for taxonomic analysis?
Various hypervariable gene regions can be used for sequencing taxonomic analysis. Selection of primer combinations that we use for the amplification of these regions is continuously expanded. You can find the current available primer sets in our Sample submission sheet.
You need to use some other primers and cannot see them in our list? - Get in touch!
Results guarantee
- For each sample that passes the initial quality control we aim to provide at least 10,000 or 20,000 reads, depending on the technology. This output is not guaranteed, however, due to unpredictable nature of every individual sample. Upon request, this output can be increased (charged).
- The sequences obtained will be sorted according to the combination of indexes into files representing individual samples and analysis of sequencing quality indicators such as count and length of sequences, phred score, %GC, etc. will be performed.
- As output you will receive data in FASTQ format divided into files according to individual samples. Upon request, the outputs can be processed in another form. Please specify this when ordering.
Data analysis
Apart from laboratory processing of your samples it is also possible to order data analysis.
Results will be provided as tables/graphs and will demonstrate semi-quantified representation of microorganisms. If you already have raw data available, you can order their analysis separately.
|
 |
|
What is included:
- The quality of raw reads is evaluated using FastQC and MultiQC.
- Adapter sequences are clipped and low-quality bases trimmed.
- Metataxonomic analysis is performed using Silva (16S/18S) or UNITE (ITS) databases.
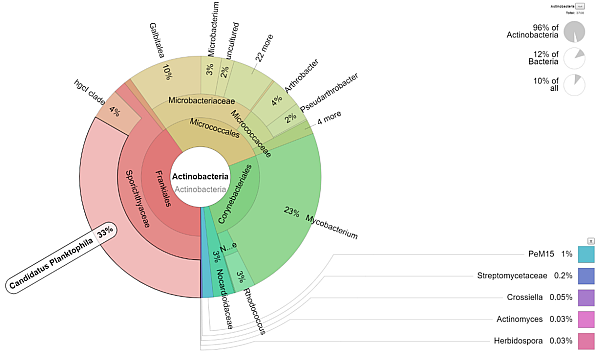
- Bar-charts of the taxonomic abundances of each category and each sample are generated.
- Additional visualization of the results is performed using Krona tools (see picture above).
|
We will provide you with demo data upon request!
Course or workshop
- If you are interested in learning how to analyze the data, visit our regularly organized workshop!
- Beginners may also consider our 2 day NGS introductory course.
Sample requirements
Follow our Sample submission guidelines.
- The samples must be delivered exactly in the concentration according to instructions! Too low a concentration could lead to a low PCR yield, a high concentration to different yields or even inhibition of the reaction (a high DNA concentration may be associated with a high concentration of inhibitors). When isolating DNA, keep in mind that more frequent washes and lower yields are certainly better than higher yields with inhibitors. Please provide samples for metagenomic analysis in strips or 96-well plates.
- It is recommended that at least one of the samples you send is a negative control. For example, pure water can be used as a suitable control, from which you “isolate” DNA using the same procedure as from real samples (because not all isolation kits are DNA-free). We always include our own positive and negative control samples.
- Please note that the success of the analysis depends very much on the integrity and purity of the DNA you provide! The use of degraded or contaminated DNA results in amplification artifacts. Contaminating substances that interfere with amplification are also to be avoided. We recommend removing RNA during DNA isolation.
How to order the service
The metagenomic sequencing can be ordered and commissioned online as a package starting from 24 samples. If you want to order an analysis of a different count of samples (eg only a few samples or, conversely, thousands), contact us to prepare an individual calculation.

 Tailed amplicon sequencing
Tailed amplicon sequencing
 RNASeq and differential gene expression analysis
RNASeq and differential gene expression analysis
 Whole exome sequencing
Whole exome sequencing
 Illumina sequencing
Illumina sequencing
 Long-read sequencing technologies
Long-read sequencing technologies
 NGS Services à la carte
NGS Services à la carte
 Instruments in our fleet
Instruments in our fleet
