
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Pacific Biosciences SMRT sequencing technology enables sequencing of fragments of both tens of kilobase and hundreds of bases in length. In the previously used PacBio RSII system there was a problem with the loading of fragments in lengths of 500 - 1000 b. For fragments shorter than about 500 b the diffusion method was used, which unfortunately allowed a maximum of 40% loading only, and moreover it showed a strong tendency to more efficient loading of shorter fragments. For fragments longer than 1000 b, the paramagnetic bead loading method could already be used effectively. However, the loading of fragments between 500-1000 b was very problematic. It is not only for this reason that the RSII sequencer has become obsolete nowadays, although it has been on the market for a short time, and its use is not recommended by our company anymore.
When loading SMRT chips of the Sequel system, which is the technological successor of the RSII model mentioned above, and on which we are currently analyzing your samples, a pre-extension step is used, which eliminates the mentioned drawbacks by diffusion. This step can also be used for libraries with up to 15 kb fragments. Longer libraries are still being loaded with paramagnetic beads. This results in a situation where similarly successful loading can be expected for libraries of different lengths and the resulting sequencing capacity is a pure multiple of the number of reads and the length of reads.
But what kind of reads does the Sequel system generate?
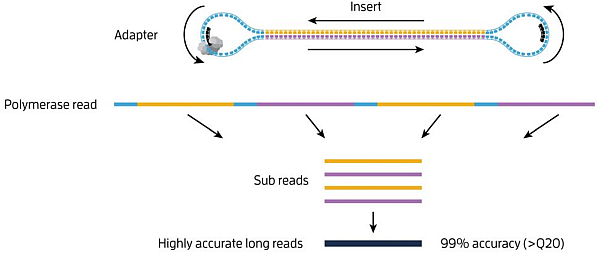
Obviously, such high reading accuracy cannot be achieved with templates of all lengths. The SMRT sequencing speed is 3-4 b/s. The maximum sequencing time is 20h. Thus, up to ~ 300 kb of polymerase reads can be theoretically achieved. However, the quality of these reads will be very poor as the polymerase read will in fact form one sub-read only.
The whole concept leads to the fact that the sequencing output in the form of a number of bases depends very much on the nature of the input material (amplicons vs. gDNA, HMW gDNA vs. degraded gDNA) and on the subsequent sample processing, where we can include a fragmentation step in order to obtain shorter but high-quality sequences, or longer ones of lower quality.
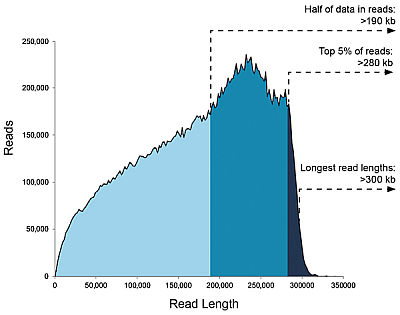
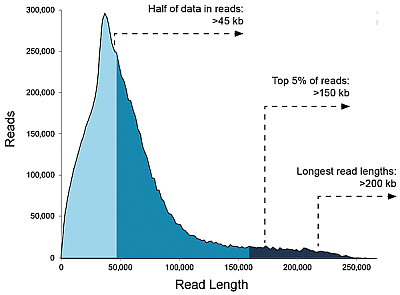
Charts below show that with shorter fragments you will achieve a larger sequencing output overall.
|
Libraries <20 kb, data/SMRT flowcell: Up to 50 Gb
|
|
Libraries>20 kb, data/SMRT flowcell: Up to 20 Gb
|
You may find it to be the other way around, but be aware of the following:
The situation is even better illustrated by the following picture:
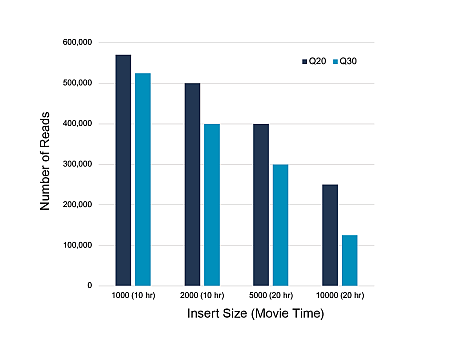
When planning PacBio sequencing, please take this into account or contact us.
Images by Pacific Biosciences
NGS Lab, ngs@seqme.eu