
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Sanger sequencing is associated with certain technical limitations, and one of the most important is read length. Typically, even with a perfectly flawless design, not more than a roughly 1,100 bases can be expected. But what is the lower limit of how long or rather short templates can be to be able to successfully sequence them? And can we sequence from the first base just after the sequencing primer? We will look at this issue in this article.
Problem description
Let's start a little from the back - if you fill out our order form for Sanger sequencing, you must also enter the length of the template and the minimum allowed length is 100 bases. This does not mean that it is not possible to sequence a shorter template. Rather, it usually doesn't make much sense, because sequencing chemistry routinely used (also in our lab) will start reading about 20 bases behind the primer. If we consider a template of length 100 b and primers 20 b, it means that you will get 60 bases from each direction and you will be happy to overlay the forward and reverse sequences.
In addition, the data just behind the primer is usually not nice. First, the shorter the PCR product, the harder it is to purify (eg on columns). The unremoved contamination present, typically salts, then interferes with the reading of especially short fragments, because they have a similar speed of migration in the capillary. And secondly, there is a certain limitation of the technology as such. The DNA fragments (sequencing reaction products) migrate in the capillary during separation depending on their molecular weight more or less proportional to their charge, which is the essence of the method. However, all DNA fragments must be fluorescently labeled (terminally, at the 3 'end, each with one fluorophore according to the terminal base), each ACGT base has a different fluorophore and these have different molecular weights. So, if we had the same DNA fragment (identical sequence) labeled with different fluorophores, they would have a different molecular weight and migration speed. In practice, we do not have two completely identical fragments with different fluorophores, but two fragments differing in base and having the same or different fluorophores. This picture shows it nicely:
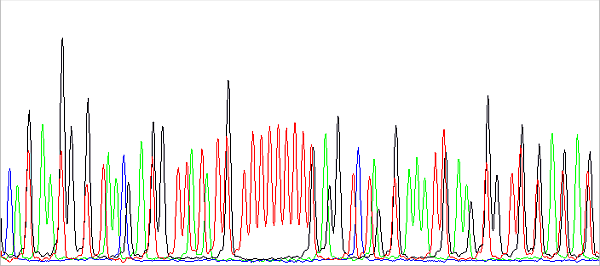
This is a preview of the raw data before basecalling. About in the middle you see a sequence of 11 red peaks (T) interrupted by a black one (G). The red peaks are evenly spaced because they all have the same fluorescent label and differ in base. Black also differs in base compared to red to the left or right of it, but has a different fluorophore (with a different Mw, in this case lower than the Mw of the red fluorophore), so it migrates in the capillary faster. In fact, it is a bit ahead, more to the left than we would expect it to be. Therefore, the sequence cannot be read from the raw data or is difficult to. Of course, this effect is removed when analyzing the data:
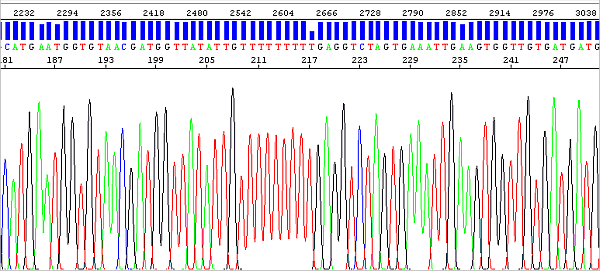
The migration of fragments (peaks) in the electrophoretic field is of course always affected by the fluorophore bound to them, and this effect is greater the shorter the DNA fragment (the fluorophore thus makes up a higher percentage of the total molecular weight). The analysis algorithm has to perform peak mobility correction differently if the fragment is say 30 or 300 bases in size, which is a bit of a challenge.
The problem of reading short templates can be solved (in part) by using alternative sequencing chemistry that can read a little better just after the primer (kits called BigDye Terminator v1.1). However, the typical requirement of a typical client is to sequence as far as possible, about 1100 bases, and this sequencing chemistry has a problem with that. You may start reading some ten bases behind the primer instead of twenty, but reading 1100 bases will be difficult. It's just something for something.
So what one can do?
There is a very nice strategy that solves this situation and combines two approaches, both feasible on the client side. You don't necessarily have to do both, one may be enough, but if you use both, it will have a greater effect.
First, you extend your PCR primers with overhangs compatible to sequencing primers. Typically, sequences for the M13 forward and M13 reverse primers are used, but in principle it does not matter. This will extend the PCR product by some 40 bases. Then you purify it (you also remove the PCR primers) and add the sequencing primer M13, which you have ordered having a specific tag (available from some primer providers, for example Generi Biotech in the Czech Republic). We've discussed these primers in detail here, so let's just briefly say that the tag allows you to read right after the primer, from the first base of your PCR product!
Below you can see an example - sequencing of a short PCR product:
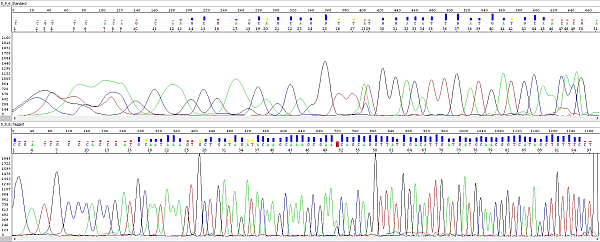
Upper electropherogram shows the trace obtained "classically", using PCR primers without M13 overhangs and a sequencing primer without the specific tag. One of PCR primers was used for sequencing too.
Lower electropherogram is the same PCR product, obtained by the strategy described above (primers with M13 overhangs) and sequenced with a tagged M13 primer. A closer look reveals that it is the same sequence (peak 25 at the top corresponds to peak 58 at the bottom). The lower sequence is not only longer, but of course also better overall and ending with the M13 overhang sequence. As we verified with the customer who sent us these samples, it really starts from the first base after the primer.
This is a completely reliable procedure. In addition, it has the advantage that you can add M13 overhangs to different PCR primers, and in fact you will always sequence all PCR products using the same M13 tagged primers.
If you decide on this strategy, we will be happy if you let us know in advance. At least because it's such a nice departure from our daily routine - we are always happy to see nice results of this type. All the more so as the above-mentioned upper electropherogram is not the result of our work but of a competing company, while lower shows our results, obtained by our recommended and described strategy after the customer lost patience with the original sequencing company. But you definitely do not have to let us know - on our side, these samples will be sequenced without any modification of standard sequencing protocols, the whole "magic" is in your hands.
Sanger lab, info@seqme.eu