
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
It is not the aim of this post to provide a detailed description of all problems you may observe when evaluating your sequencing results. Instead, I focus on most frequent problems and recommend some steps to take for your consideration. Additionally, I am not covering issues related to instruments and sequencing reagents used in our (or any other) sequencing lab because first it is our responsibility to secure a problem-free sample processing on the instrument and second, from the user‘s point of view, it is of low interest because you cannot influence it anyway.
For simplicity, the text is divided into three chapters named after three major symptoms observed. Their troubleshooting, however, is not always straightforward and as I am trying to explain at least some symptoms may have different causes. First I deal with weak or no fluorescent signals, then with good signal (at least at the beginning) but unacceptable results because of declining peak intensity and the last chapter covers “too many peaks” scenario.
Let me show an example first:
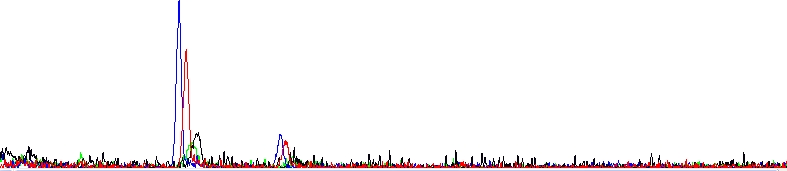
Absolutely dead sequencing trace
As you can see, there is clearly nothing, only two dye blobs. The reaction failed.
Possible explanations in this case are: template DNA was not added at all, is not of sufficient quantity or is severely contaminated with some sequencing inhibitors. The sequencing primer was not added at all, is not of sufficient quantity or does not bind the template.
It is helpful to know that if the reaction fails for whatever reason or its signal is very weak, it often has one or two large peaks (sometimes called Dye blobs) visible in the raw and analyzed data. This is residual amounts of dye terminators that were not removed when the reaction was cleaned up.
We at SEQme always clean reactions in the same way, however, the cleanup is not typically 100% successful if the dye labeled terminators were not consumed at all in the reaction to synthesize any products and consequently are nearly impossible to remove completely, simply because there is too much of them. Hence, the blobs are not a mistake themselves, just a consequence of it. One could also say that observing dye blobs in your electropherograms may suggest a low template concentration.
Depending on what is wrong, relevant corrective measures can be applied but frankly speaking – weak or no signals are a bit difficult to troubleshoot. You can typically see the reaction failed but possible causes are plentiful. One can compare this to your TV malfunction – it might be the TV itself (and its hundreds of parts), your remote control, antenna or even an electrical grid. And please help yourself…
First, make always sure you follow our recommendations concerning template concentrations required. Using too little DNA is a mistake leading to no or weak signals but using too much is a mistake too!
Please note that a spectrophotometer is not necessarily a reliable mean of measuring DNA concentrations. Primers, proteins and RNA absorb at 260 nm and can give rise to artificially inflated estimates of the DNA concentration. We strongly recommend agarose gels. If you use a DNA ladder to compare to, you can get a much more reliable estimate of DNA concentration of your samples and information on their quality as well. Also, note that Nanodrop is a spectrophotometer too (not a surprise probably to most readers). If you do not want to run a gel we recommend Qubit.
Overall, accurate estimation of DNA concentration is critical for the sequencing reactions to function properly.
Assuming your DNA concentration is ok but you observe no signal you better check the amount of sequencing primer. A common error is that primer concentrations are off by a factor of 10 or 100. If only reactions with certain primer fail, DNA may not have a site for the chosen primer because of a mutation in the primer binding site.
So once again, assuming your DNA concentration is ok and you have no reason to blame your sequencing primer, one has to conclude some DNA sequencing inhibitors are probably present in your sample.
Having enough template is a mandatory but not sufficient prerequisite for successful DNA sequencing. Its purity is equally important. Sequencing reaction is quite sensitive to various inhibitors, even buffers are usually not recommended to dissolve your DNA (use HPLC-grade water instead).
Common and frequent inhibitors are salts, leftovers from template purification kits (e.g. ethanol or traces of various buffers typically containing high concentration of salts), dNTPs (!) etc. Additionally, there are other impurities which do not kill the reaction but should be avoided as well – namely PCR primers and non-specific products.
One should therefore pay special attention when considering how to clean up templates. In principle, you are presented with a choice between three options, depending on what you see on the agarose gel.
dNTPs can cause a lot of troubles
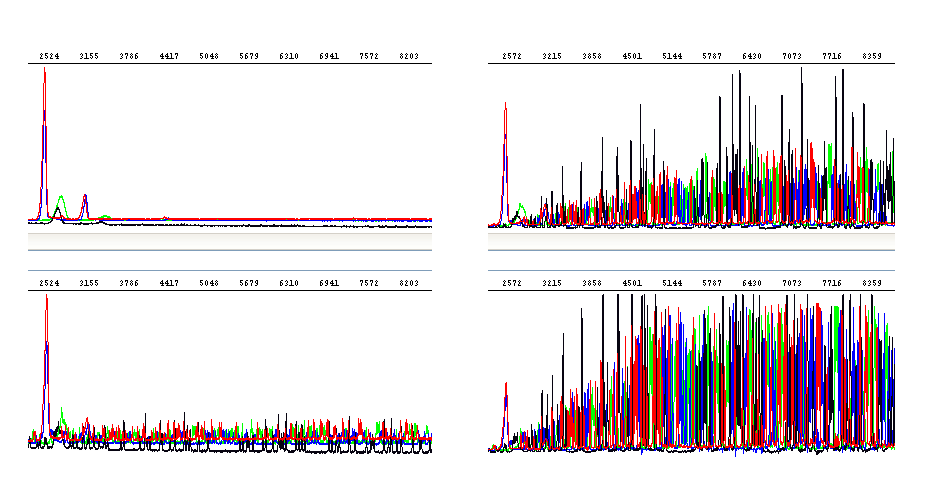
Weak fluorescent signals can also be interpreted as high background. What you observe is in principle a clean and nice sequence, nevertheless peaks are relatively low (check raw data) and the underlying background significant. The most probable explanation is carryover of dNTPs from the PCR.
The presence of any residual amounts of the dNTPs will interfere with the sequencing reactions. The residual dNTPs will change the ratio between the dNTPs and ddNTPs in the sequencing mixture and reduce the incorporation of the fluorescently labeled ddNTPs that allow one to visualize the sequencing reaction products. In other words, the residual dNTPs will reduce the strength of the peak signals even though there was sufficient DNA and primer. Surprising this may be, the carryover of dNTPs from the PCR step will definitely do no good to the sequencing reaction.
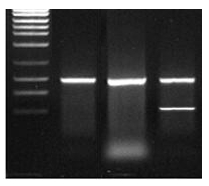
The product is viewed as a single clean band of the desired length
The simplest and most efficient method of cleaning is the Exo/SAP mixture (USB or homemade). Exonuclease I degrades single strand primers and Alkaline Phosphatase dephosphorylates residual dNTPs. It is quick, not laborious and no sample is lost.
If using Exo/SAP for cleanup of your PCR products, we do not recommend to follow the standard protocol (2 µl of Exo/SAP per 5 µl of PCR product incubated for 15 min at 37ºC and heat inactivated for 15 min at 80ºC). It is expensive and not efficient because too fast. You should better use 1 µl of Exo/SAP per 10 µl of PCR product and incubate 30–60 min at 37ºC. Also inactivation step can be extended.
Make sure you do not use excessive concentrations of primers and/or dNTPs in the original PCR reaction (“200/200 rule” – primers: not more than 200 nanomolar each, dNTPs: not more than 200 micromolar each). At higher concentrations not all primers and dNTPs are removed and the downstream sequencing reaction can be affected. More primers = higher background or mixed sequences, residual dNTPs = weak signals (see above).
The product is viewed as a band of the desired length but unfortunately there are other products too, relatively short, including but not limited to primer dimers
Using Exo/SAP mixture is out of the question. The most common approach would be a PCR purification kit and loading of the templates to DNA-binding spin columns. These columns must have an appropriate cut off limit allowing removal of small fragments and keeping only the desired template. PCR (and/or plasmid) purification kits are very common, available from various vendors and easy to use. But some of spin columns may not have a really favorable shape and this has a direct influence on the success of purification when spinning them in angle rotors (as everybody does).
The penultimate step of the cleanup procedure is spinning empty columns, a minute or two, to dry them out. Then an elution buffer is added and after another brief centrifugation samples are collected and shipped for sequencing. But they may still contain some ethanol or salts from buffers used during purification. These should have been removed when centrifuging empty columns but recommended spin times are usually too short and due to the shape of spin columns some liquid remains in the cup and is removed not sooner than at the elution step. As a result, your sample gets contaminated.
You can confirm this easily. Proceed as described in the protocol but just before elution transfer the inner cup into a clean eppendorf tube and spin empty for 5 min at recommended speed. In most cases you will see a small volume of some liquid at the bottom of the collection tube, most likely ethanol (up to 10 µl). This liquid will inhibit your sequencing reaction.
What can be done here? The simplest way is to extend the centrifugation time to approx. 3–5 min when drying. But there are inner cups with internal lip holding small (dead) volume of liquid when centrifuged in angle rotors. It is therefore recommended to centrifuge empty tubes for 2 min to dry them out and then, without removing the tubes from the rotor, rotate the inner cup 180º and spin again for additional 2 min. This way any liquid trapped inside should be removed. Make sure the inner cup is completely dry and you cannot see any traces of liquid and transfer it to the elution tube. Proceed with elution as recommended by the kit manufacturer.
The product is viewed as a mixture of bands of both the desired and undesired length, relatively long
Only gel purification can solve your problem. But you may rather consider optimizing your PCR reaction to get rid of unspecific products because gel purification is laborious and samples obtained are in many cases inhibited as in the case of column purification described above.
Sanger lab, info@seqme.eu