




 Metagenome sequencing
Metagenome sequencing
 Tailed amplicon sequencing
Tailed amplicon sequencing
 RNASeq and differential gene expression analysis
RNASeq and differential gene expression analysis
 Whole exome sequencing
Whole exome sequencing
 Illumina sequencing
Illumina sequencing
 Long-read sequencing technologies
Long-read sequencing technologies
 NGS Services à la carte
NGS Services à la carte
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl

In case of specific requirements, we can provide sequencing using also other sequencing platforms
Last update: February 1 2024. For those interested in NGS technology we recommend our regular two-day course! More information here.
Currently we offer processing of your samples using following technologies and instruments. Given the rapid technological developments, it is possible that despite all efforts, the information provided below is not 100% up to date.
|
|
NovaSeq X Plus and NovaSeq 6000 (Illumina)We can offer chips (flow cells) having various outputs of sequencing data in terms of read length and amount (sequencing capacity). Using the proper settings and data allocation (see ShareSeq) it is possible to ensure economic use of the device meaning with one type of technology, experiments with different sequencing capacity requirements can be realized. It is possible to carry out a pilot experiment with a lower capacity and then design a larger scale experiment according to the results. |
|
|
|
|
|
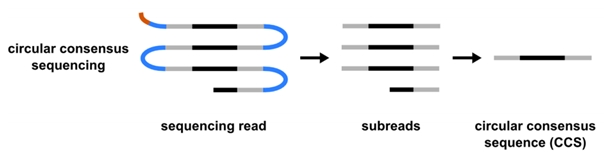
PacBio Sequel IIe and Revio (Pacific Biosciences)For special applications (for example de-novo genome assembly or when sequencing amplicons and repetitive motifs) longer sequences need to be obtained and SMRT technology used in PacificBiosciences devices can provide this. Compared to other technologies, there is no template amplification step during the preparation of the sample for sequencing and therefore some epigenetic modifications can be recorded directly. A circular DNA molecule serves as a template and DNA polymerase reads it until the sequencing run is finished. Fragments of up to a few tens of kb are therefore repeatedly read and by comparing individual readings performed by the device automatically, the correct identification of every base is achieved. This strategy is called "circular consensus sequencing". By reading 8-10x, reading accuracy is improved to more than 99.99%, which is comparable to that of Illumina.
(a picture by PacificBiosciences, adapted)
Note: If you are interested in sequencing by using this technology, please contact us to receive a quote tailored to your project. |
|
|
|
|
|
MinION / GridION (OxfordNanopore)A very interesting option for applications requiring long reads are the Oxford Nanopore sequencers. Oxford Nanopore technology has historically struggled with a reputation for poorer sequencing quality. Today, however, it can be said that this problem is a thing of the past, because when using optimal basecalling algorithms, the data is of perfect quality. We recommend the Oxford Nanopore technology for metagenome sequencing! |