
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Intro
Although state-of-the-art Next-Generation sequencing (NGS) technologies generate an adequate amount of sufficient quality data for de novo genome assembly of organisms for which the reference sequence is not known yet, the genome assembly process itself is still a major challenge for bioinformaticians. In this article, we will try to outline the issue of assembly and show possible solutions.
One of the most widespread NGS platforms is Illumina. The main advantage of this platform is the high quality of reads, the ability to sequence paired end reads, the relatively affordable cost of sequencing and robustness. The disadvantage is the reading length, which is limited to 2x300 b for this platform. Even with high coverage there are specific regions of the genome which are insufficiently, or not at all, sequenced. We are talking about GC rich regions, chromosome beginnings and ends, repetitive regions, etc. These issues may have negative effect on the process of assembling the complete genome, which results in incomplete and fragmented sequences and leads to the formation of a larger number of shorter contigs, which is not easy to properly assemble into one coherent unit. One way how to bridge these inconsistencies is usage of mate-paired libraries. This kind of sequencing does not fill the gaps in assembly but helps to arrange contigs to the correct order.
Another possibility is to incorporate long fragments of DNA obtained from different sequencing platform into assembly. Oxford Nanopore Technologies (ONT) seems to be a suitable adept. According to the manufacturer's specifications, it is possible to obtain reads of up to several megabases. Ordinary sequencing run contains shorter fragments (few kilobases) which are, however, sufficient for purpose of analysis. Even if the ONT lags behind Illumina in quality, it is being very quickly developed and improved.
One could certainly make De novo assembly based only on long reads but in case of higher error rate higher read depth is necessary. Here the question arises – is it possible to combine these sequencing technologies and obtain the highest quality genome? The answer is yes and we have more than one solution. For example, hybrid assembly is able to combine shorter high quality reads from Illumina with long reads which are ideal for filling the gaps between contigs. Second possibility is assembly based on long reads (e.g. ONT) and improved with the high quality Illumina short reads - so-called polishing.
We have recently addressed a similar situation - we performed de novo assembly of the bacterial genome using data from both Illumina and ONT. The reason for assembling the genome using multiple approaches was to try to find the most ideal assembly method for the data.
SPAdes Genome Assembler was chosen for hybrid assembly. This program uses de Bruijn graphs for read assembly and was developed for de novo assembly of small genomes. SPAdes enables to combine short reads and long reads or assemble them separately. Second approach was assembly of long reads using Canu assembler. This assembler focuses on analyzing reads from PacBio and ONT platforms. Canu works in three steps: corrections, trimming and assembly. Inconsistent parts of assembly of long reads are repaired with Illumina data in following step calls polishing.
Note: All programs used in this comparison are free and can be distributed and/or modified according to the GNU General Public Licence version 2.
Data
In our case, the new bacterial genome was assembled with estimated size 3 Mb and higher level of GC content. For this purpose were prepared two libraries.
For comparison, four different approaches of assembly were run:
Assembly results were compared by program Quast. Only contigs longer or equal to 500bp were included in the statistical calculations. See summary in the table below.
Results
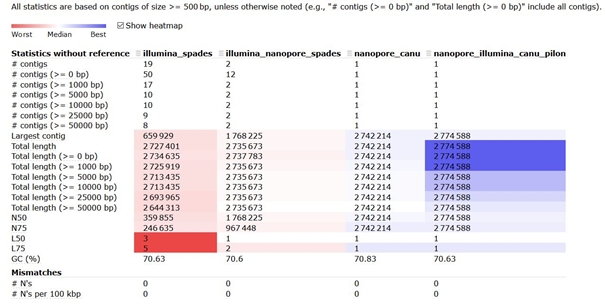
Statistical comparison of all assemblies - Quast
Comment
Total length of assembly was around 2.7Mb in all four examples which corresponded to the expected size of studied genome. In the first case (Illumina_spades), the genome was assembled into several contigs. More than half of them were shorter than 500 bp. The longest contig covered about one quarter of the total length of assembly. In comparisons with the second analysis (Illumina_nanopore_spades) interesting difference can be seen between assembly composed only from Illumina data and hybrid assembly. In case of hybrid assembly, count of contigs dropped down (nevertheless, majority of contigs was shorter than 500 bp). Total length of assembly is slightly longer, but more interesting is the size of longest contig which grew up more than 2,5 times in comparison to the assembly from Illumina data! It is obvious that long reads helped to bridge parts of genome which could not be properly assembled with short reads.
The assembly of long reads produced one final contig (nanopore_canu). Compared to the previous results, we can observe a slightly higher GC content, which may be due to the poorer quality of ONT reads. The last column represents the results of the assembly of long reads (Canu) after the subsequent correction by Pilon using Illumina reads (Nanopore_illumina_canu_pilon). This correction helped to slightly lengthen the final contig and adjust the GC composition.
These results clearly indicate that the de novo assembly created by combining short reads (Illumina data) with long reads (ONT data) contributed greatly convincing results and helped to technically improve the final assembly of the genome. Although it is not possible to say with 100% certainty that this genome was assembled completely and correctly (which, unfortunately, can never be done in principle), we are close to the expected assumptions and commonly used metrics in this case clearly indicate the suitability of this approach. In general, it cannot be declared that the only method of assembly with subsequent polishing is the best approach. Subsequent genome annotation and in vitro control of results are necessary. It is always necessary to take into account the specifics of individual species, the overall size and complexity of the genome. Therefore, we recommend using multiple procedures during data analysis. The most suitable approach for specific data should be chosen based on critical evaluation of results.
It is certainly clear that this combination of ONT and Illumina technologies, inevitably more costly than the use of only one of them, offers fundamental advantages for successful de novo assembly. If you are planning a similar project, we recommend that you contact our application specialists soon enough. The above example is proof that the basis of success is to plan the project well and we will be happy to help you!
NGS Lab, ngs@seqme.eu
Literature