
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Illumina sequencing technology is currently dominating the market. It offers a number of advantages over other technologies, in particular high sequencing output and accurate reading, but unfortunately this does not mean that it is completely free of imperfections. One of its main drawbacks is, in particular, the reduced efficiency of sequencing of templates bearing a uniform sequence motif and the consequent risk of devaluing the entire sequencing run by improperly sequencing such templates.
For example, the following templates and libraries created from them may have a problem:
Another drawback may be the improper selection of index combinations when designing a multiplex, since the index is read de facto as a separate read and as for sequence diversity, the same rules apply to it as to the insert sequence.
If this happens, what really occurs and what are the solutions?
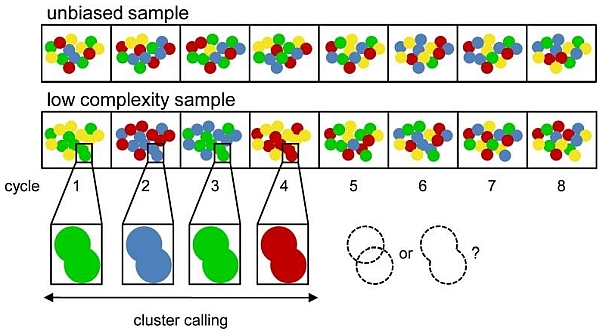
When a uniform sequencing motif is present, too many clusters on the sequencing chip emit a similar signal. This complicates the definition of clusters (i.e. cluster calling) when sequencing starts, as shown in the figure (adjusted by Krueger F et al., 2011):
In addition, the sequencer detector is “blinded” by this uniform signal, as shown in the following figure (based on Illumina materials):
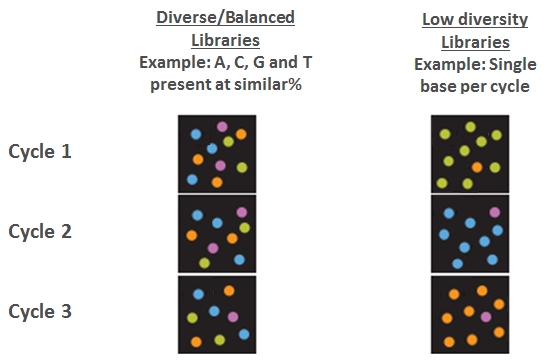
The situation worsens even more when sequencing the second read in paired-end sequencing, when the diameter of the clusters increases and their boundaries get closer together.
There is a solution, of course, but it is at the expense of one of the main strengths of the technology - it is necessary not to have such high expectations regarding the amount of data we want to obtain. In practice, we need to reduce the concentration of the library applied to the chip, so that the density of the clusters on the chip is smaller and the clusters are sufficiently spaced. Furthermore, it is necessary to dilute these uniform sequences with a library of fragments that have the most varied sequence over their entire length (the sequence of this library is then completely removed from the dataset obtained). These two options are universally valid.
A specific way of avoiding this situation is in the case of amplicons (with inline barcode or specific overlap), where we can design primers containing the so-called "heterogeneity spacer", see figure below adapted from Douglas et al. 2014: At the 5 'ends of specific primers for the V3-V4 region of the 16S gene these spacers are included:
![]()
All of these measures are used to an extent that depends on the uniformity of the sequenced library. In extreme cases, their combination can lead to a 50% reduction in sequencing output, but if neglected, the entire sequencing run may be degraded. This is particularly important in paired-end sequencing, when the diameter of clusters increases and their boundaries are getting closer together. The quality of the first read may still be satisfactory, but the quality of the second read may be lowered significantly, as shown in the figure below.
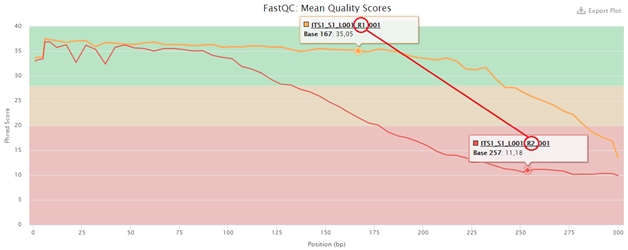
In summary, although templates of this type pose a challenge to Illumina technology, it is a challenge that can be solved (mostly) at the sequencing laboratory level. We emphasize, however, that in these situations you cannot expect the technical specifications of the manufacturer regarding the amount of data you will get to be met, as they are, of course, defined on the basis of libraries with high sequence diversity.
Note: This and other related issues are solved in detail at our courses.
NGS Lab, ngs@seqme.eu
Literature:
Douglas WF, Bing M, Gajer P, et al.; An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform; Microbiome 2014; 2:6
Krueger F, Andrews SR, Osborne CS; Large scale loss of data in low-diversity illumina sequencing libraries can be recovered by deferred cluster calling; PLoS One 2011; 6(1):e16607