
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Clients of our sequencing laboratory often require the preparation of sequencing libraries using rRNA depletion (ribodepletion), ie including rRNA removal. As we sometimes find that the results of rRNA removal are unsatisfactory, we performed tests described below. Their goal was to provide a more detailed explanation of where the catch is in the case of rRNA-depleted libraries, and at the same time to provide some recommendations to you, our customers, on how to prepare total RNA for this type of analysis.
Introduction
Ribosomal RNA (rRNA) is the most abundant component (> 80%) in total RNA isolated from cells or tissues. However, for many applications, rRNA is irrelevant. Its sequencing unnecessarily increases the required sequencing capacity and, ultimately, the cost of laboratory and data analysis.
Thus, in the first step of preparing RNA-seq libraries, it is more than desirable to get rid of this unnecessary rRNA. There are currently several protocols available that use different strategies for this purpose. These methods naturally have their strengths and weaknesses.
The most common form of sequencing library preparation begins by enriching for mature poly-A transcripts by hybridization to oligo- (dT) magnetic beads. Subsequently, only these specifically bound fragments are used to prepare the RNA-seq library. In plain language, we only "pull out" from the total RNA for further processing those molecules that have a poly-A end. The advantage of this approach is that sequencing libraries are created on the basis of transcripts encoding proteins, which is ideal, for example, for differential analysis of gene expression. This method of so-called poly-A selection is used for most transcriptome studies also because the required depth of sequencing is relatively low.
However, sometimes this method fails. One of them is species compatibility - poly-A selection can only be used in eukaryotic organisms where polyadenylation takes place. It is therefore not applicable, for example, to bacteria. It is also important that in the case of poly-A RNA-seq libraries, the quality of the data is significantly affected by the integrity of the total RNA. If the input material shows a higher degree of degradation, significant data bias towards the 3 'region may result. And last but not least, using this method, we are only able to sequence RNA bearing the poly-A end, so we do not get information about immature pre-mRNA, non-coding transcripts and all other RNA molecules not having a poly-A end.
The second method for removing rRNA is to use specially designed capture probes that specifically hybridize to rRNA (we use DNA probes). The rRNA thus bound is then removed magnetically or enzymatically and the remaining material is used to prepare an RNA-seq library. Again, in plain language, for the need of further processing, we will "throw out" the rRNA molecules from the total RNA. We are talking about so-called ribodepletion.
Although it is necessary to take into account that in some cases up to twice the sequencing capacity is required to achieve the same level of detection sensitivity for gene expression analysis, this method has some significant advantages. rRNA depletion offers an attractive option for simultaneous detection of coding and non-coding RNAs: mRNA, pre-mRNA, and a variety of other functionally relevant short and long non-coding transcripts. It therefore provides significantly more information than simple poly-A selection. Another advantage may be that rRNA depletion methods can be used in certain circumstances to sequence RNA samples with lower integrity (but of course, any degraded RNA may adversely affect the output and reliability of the assay). For possible analysis of such problematic samples, we would therefore rather recommend the QuantSeq protocol.
In general, in the case of a well-performed ribodepletion, the rRNA will represent 5-10% of the resulting data. The success of ribodepletion, ie the removal of rRNA, depends on a number of factors.
The success of ribodepletion
The first important prerequisite for successful ribodepletion is the use of the proper reagents compatible with the target organism. In this respect, it is possible to use pre-designed commercial probes (eg Human / Mouse / Rat, Bacteria) or it is possible to design your own set of probes for a specific organism (here, however, it is necessary to take into account higher costs and more time required). In this context, it is worth noting that in the case of plants, a reliable and universal set of probes is not currently available and it is always desirable to test or optimize probes for a specific plant species and type of plant tissue.
Another fundamental prerequisite for the success of ribodepletion is the sufficient quantity and, of course, the quality of the input RNA sample. While the concentration and integrity of total RNA can be verified quite easily, the detection of other parameters is more difficult. We are talking in particular about the various inhibitors that may be present in the RNA sample (especially salts, detergents, alcohols, enzymes, etc.) and which may play a crucial role in the success of ribodepletion, while it is essentially impossible to say with certainty whether or not they are present. On the one hand, these substances may prevent the successful hybridization of ribodepletion probes to rRNA, resulting in imperfect removal of rRNA from samples, and / or may inhibit ongoing enzymatic reactions. An example is the dependence of ribodepletion success on the concentration of specific salts, where their suboptimal concentration or the simultaneous presence of other undesirable salts can greatly affect the required ionic balance.
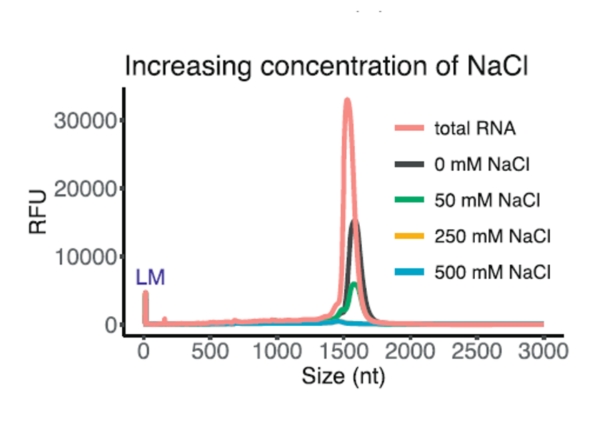
(from Kim et al., Efficient depletion of ribosomal RNA for RNA sequencing in planarians, BMC Genomics (2019) 20:909)
Last but not least, the presence of gDNA in the sample can be a problem, so it is desirable to completely remove the gDNA. If this procedure is performed using DNase I, it is absolutely critical to inactivate the enzyme completely after the reaction - due to the possible residual activity of DNase I, the single-stranded DNA probes necessary for rRNA depletion will degrade.
The problem with these analyzes is that if, for example, inhibitors of hybridization (and thus ribodepletion) are present in RNA, we will never know for sure during library preparation. We perform ribodepletion, then prepare a sequencing library, and only after it is sequenced, we can say with certainty whether ribodepletion was successful or not. But at that moment, all the money for the analysis is spent and the result is sometimes a waste.
Purity means quality
In our laboratory, we therefore focused on this issue. We analyzed a problematic test sample of human total RNA and focused on its quality (purity) and the subsequent effect on ribodepletion efficiency. In all cases, the same starting total RNA was used, but before the ribodepletion itself and the sequencing library preparation itself, this total RNA was processed as follows:
The RNA samples thus prepared were depleted of rRNA by an enzymatic method. We have preliminarily verified its success using sensitive capillary electrophoresis. The result shows that most of the rRNA was removed in samples A and B, but not in sample C.
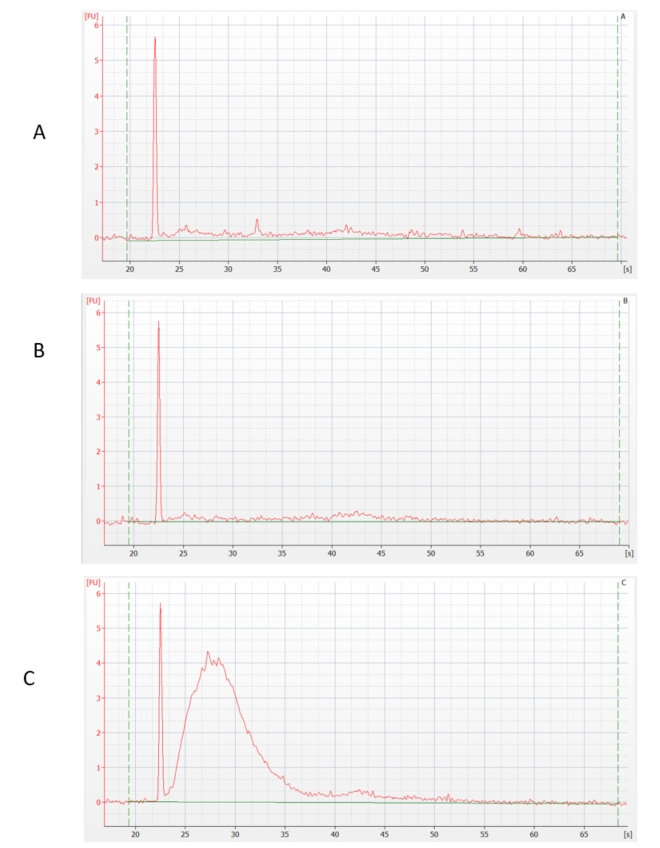
RNA concentration was also measured. The loss of RNA after ribodepletion was: sample A - 80%, sample B - 90%, sample C - 0%, which confirmed the previous statement. (These verification steps are not performed by default when preparing rRNA-depleted libraries, as the amount of material being handled does not allow this - an above-standard amount of total RNA was used for this test purposes.)
Subsequently, sequencing libraries were prepared and sequenced routinely. The obtained raw data were mapped using the bwa program (alignment using Burrows-Wheeler transformation; v0.7.17-r1188) based on the BWA-MEM algorithm in default settings against the SILVA rRNA database (release 126) and at the same time against the human cDNA reference sequence (GRCh38) with the following results:
| Sample | Count of reads mapping to rDNA (SILVA) | % rRNA in the sample |
| A | 121379 (out of a total of 2260692) | 5,3 |
| B | 119405 (out of a total of 2150301) | 5,5 |
| C | 1725514 (out of a total of 2153394) | 80,1 |
Thus, for samples A and B, which were purified before processing, the rRNA content in the data was only about 5%. This number can be considered a successful ribodepletion. In sample C, which was presumed to have non-specific inhibitors, the rRNA content in the data was 80%. The efficiency of ribodepletion is therefore more than unsatisfactory for this sample.
The resulting data therefore agree with our assumption that the presence of unspecified inhibitors in sample C negatively affected the success of ribodepletion and fundamentally devalued the experiment.
Summary
In conclusion, the purity of the RNA samples delivered to our laboratory for this type of analysis is fully under your control! Therefore, we highly recommend that you clean the samples really well before sending them to our laboratory, preferably with AMPure XP beads. This will avoid an unpleasant surprise in the form of a very high proportion of rRNA in the data after we deliver results. However, you must take into account that the loss of input material during purification can be up to 40% in our experience.
NGS Lab, ngs@seqme.eu