
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
The technology of next-generation sequencing produces huge amounts of data compared to Sanger technology. Its volume naturally depends on the design of the experiment, but primarily on the output capacity of the instrument. In principle, it is always necessary to deal with the transfer of large amounts of data into a form which enables their effective processing to allow a deeper analysis of the sequences obtained which is the very aim of the experiment.
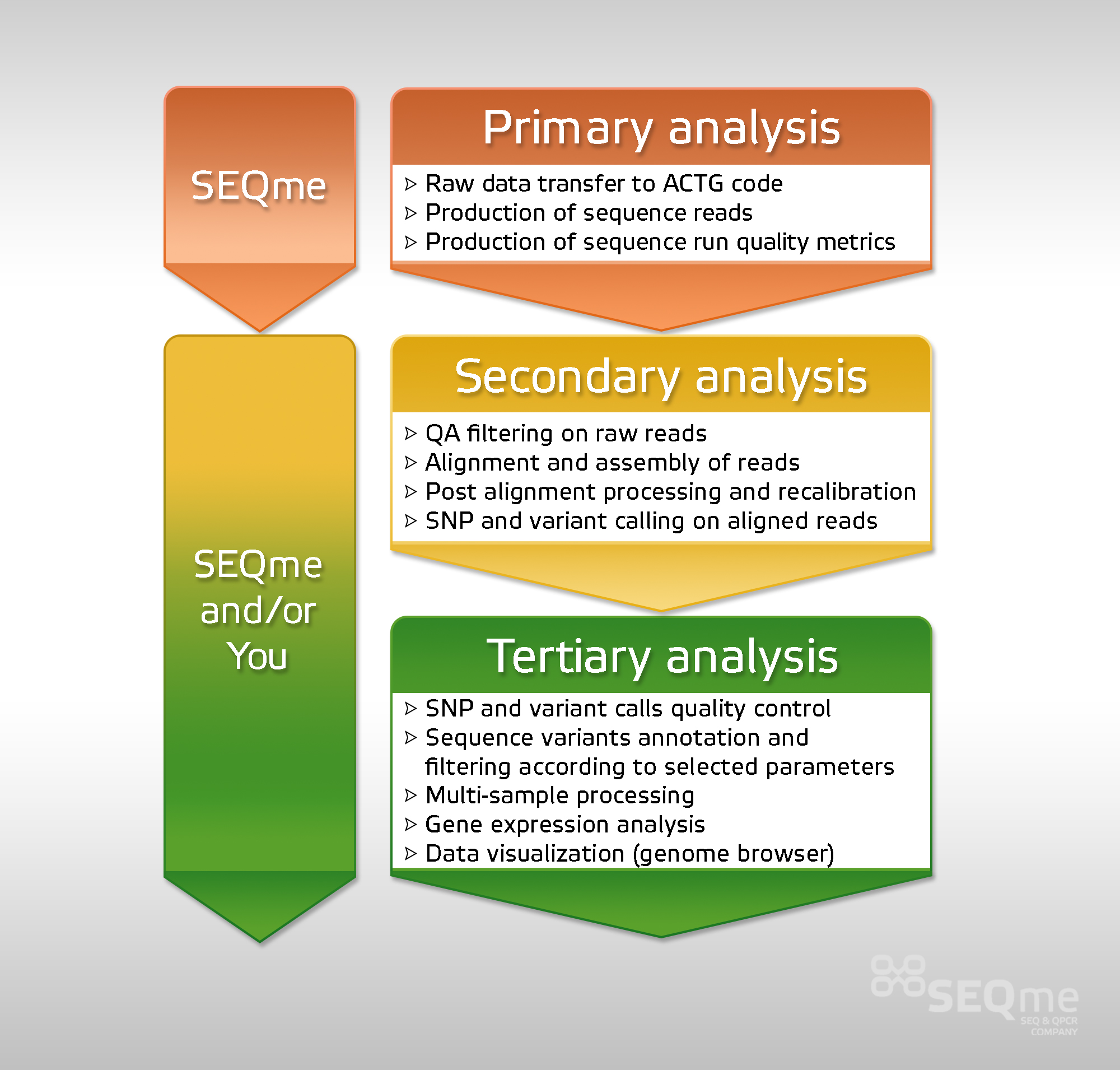
Data analysis can be formally divided into three levels:
Primary Analysis
The primary analysis consists of steps leading to the transfer of the signal generated by the sequencer to the ACTG code and to the calculation of the quality indicators for the data concerned. These steps are specific to each instrument. The output is usually a FASTQ file in combination with information about the data quality (Phred, guarantee of correct reading, in principle) for each basis. The primary analysis is performed by the sequencer and cannot be intervened in by the user.
Secondary Analysis
After obtaining raw data from the instrument (see primary analysis), the next step is the arrangement of the obtained fragment sequences (alignment of reads) and their assembly in longer sequences based on the fragment overlaps (assembly of reads into contigs and scaffolds). These steps are preceded by filtering data so that the secondary analysis works with data of sufficient quality.
The process of secondary analysis is much less demanding when using the reference sequence, where reads are assembled not only on the basis of mutual overlapping, but also according to the conformity to the reference sequence, taking into account possible differences (sequence variability), whose analysis is usually the aim of the experiment.
The next step of the secondary analysis (sometimes assigned to the tertiary one) is the detection of variants. This is the identification of differences between the acquired and reference sequences. These differences may be found at the level of individual nucleotides (substitutions, shorter insertions and deletions, “InDels”), or there may be more extensive structural changes (transversions, translocations, copy-number variations).
The secondary analysis can be conducted using appropriate algorithms and bioinformatic tools that have been experiencing continuous development on the one hand, but on the other hand have been used for so long that they have reached the point where their use is standardised. For example, Velvet (EMBL-EBI) tool can be used for de-novo sequencing, which makes use of Bruijn graphs for the assembly of genomic sequences. If a reference sequence is available, algorithms derived from the Burrows-Wheeler algorithm (BWA) are used, for instance, which provide high stability in terms of speed and accuracy.
These and other methods of secondary analyses are often arranged in a “cascade” used for one particular kind of analysis, wherein, however, the possibilities of variable configuration and tuning for other applications are retained. The whole process can be fully automated or it can be adjusted and modified. Most of these methods are freely accessible and can be applied with some advanced knowledge of bioinformatics and work with command lines or using freely available server applications. A comprehensive solution are commercial software packages from different manufacturers targeting at the secondary and/or tertiary analysis (CLC bio, Partek, etc.).
The secondary analysis is performed by the sequencing laboratory, that is us, or you as agreed in the specific project. Its output are typically standardized formats BAM and VCF, where BAM is basically alignment of reads to the reference sequence and VCF contains information on the detected variants.
Tertiary Analysis
Tertiary analysis is the final stage of data analysis and provides outputs on the basis of which scientific conclusions resulting from the experiment may be drawn. The primary and secondary analyses are essential for this last step, but they are actually “uninteresting” in terms of interpretation of the results of the experiment.
The tertiary analysis is highly variable, depending on the study in question. It is a deeper analysis of the obtained sequences and comparison of the results of several samples. The process is not usually automated and standardized, since the processing varies according to application. For example, de-novo sequencing will be processed in completely different manner than a re-sequencing study, where deviations and variations from the reference sequence are searched for. Successful tertiary analysis naturally requires well-thought-out design of the experiment (number of samples, positive and negative checks, replicates, depth read, etc.).
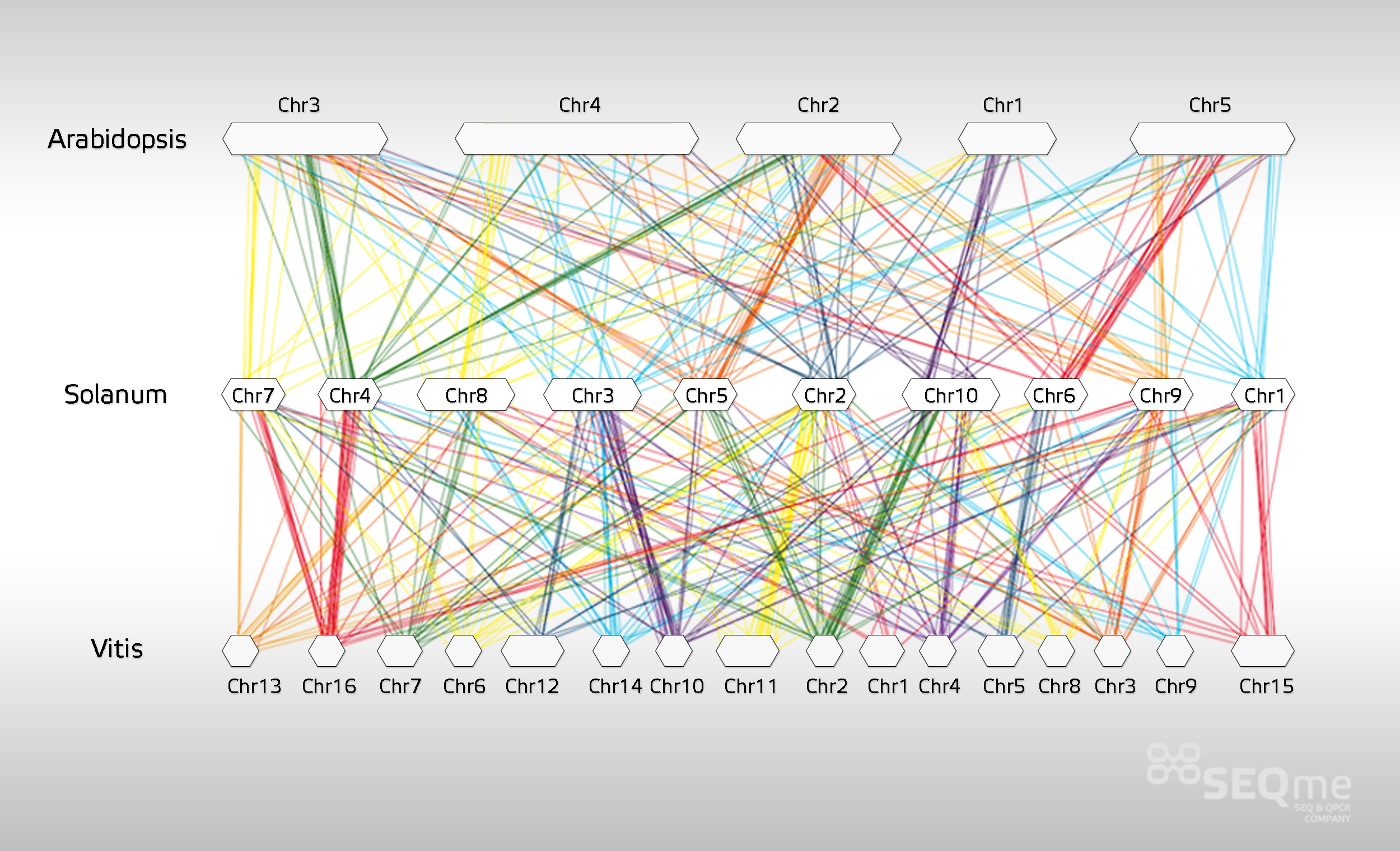
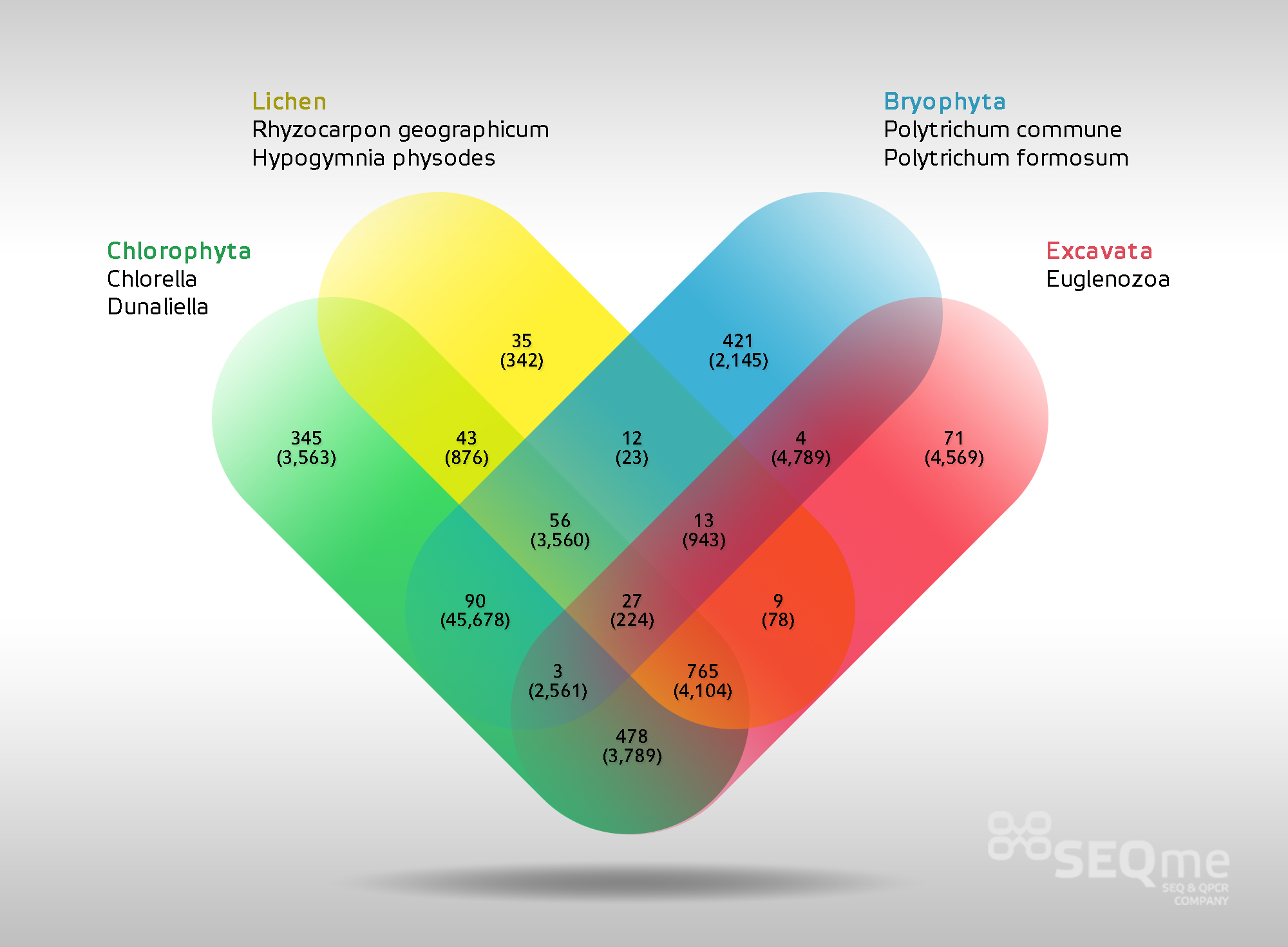
Examples of tertiary analysis outcomes
The execution of the tertiary analysis also requires suitable software with effective user interface which allows processing of the results and their visualization. The input data is usually provided in BAM and VCF format. As the tertiary analysis is not as hardware demanding process as the previous phase, the results of sequencing of entire genomes can be further processed at a slightly above standard computer. The data obtained by the analysis of exomes or targeted re-sequencing are even less demanding.
An example of tertiary analysis is the analysis of rare sequence variants. The first step is the categorization of sequence variants with low and rare frequency. If you do not have a rich enough sample, the sequences can be compared with the catalogue of sequences from the 1000 genome project or another external catalogue, such as dbSNP 129 (dbSNP 129 database is often considered the last “pure” dbSNP catalogue without many rare and unconfirmed variants of other large-scale projects). The next step is the search for the regions where the load of rare variants is highly correlated with the focus of the study. Rare sequence variants may be of different characters - neutral, protective and destructive (mutation), The analysis of variants can be performed in terms of their presence in (non)coding regions, in terms of their character (synonymous and non-synonymous), in terms of their influence on the action of protein (prediction of protein action - programmes such as SIFT and PolyPhen2), etc.
NGS Lab, ngs@seqme.eu