
© SEQme s.r.o., 2012 - 2024. All rights reserved.
Disclaimer.
webdesign Beneš & Michl
Short-read technology is at its maximum
Illumina sequencing technology is commonly referred to as short-read technology. In practice this means that it typically produces reads of hundreds of bases in length. This is, of course, relatively few and certainly does not meet the needs that we perceive among our customers. Therefore, although Illumina still dominates the market and certainly will, we also offer alternative sequencing technologies that have reading lengths several orders of magnitude higher, so they do not suffer from this drawback (and suffer from others).
If in the future some other technology pushes Illumina out of the the market leader position, it will most likely be due, among other things, to the length of reads. In 2018, Illumina attempted to "solve this technological problem" by taking over another strong DNA sequencing firm, Pacific Biosciences, which enables significantly longer reads on its sequencers (you can learn more about the advantages of each technology at our courses). But as you may have noticed, it was announced in early 2020 that both companies were withdrawing from this plan, mainly because of antitrust authorities.
2x300 b on MiSeq
It is quite obvious that if you want to sequence on Illumina, you will have to keep up with its current technological maximum in terms of read length, and this is a kit for the MiSeq that allows reading up to 600 b (2x300 setting). Below we briefly summarize our experience when using this popular setting so that you, our clients, know what to expect in this case. (Incidentally, one can conclude from the attempt to acquire Pacific Biosciences that Illumina technology probably cannot be expanded further in terms of longer reads in the near future.)
Of course, this sequencing setting 2x300 works, but it is important to say that only with a certain sample type. The main problem is that the outputs are calculated according to Illumina's specification for an optimal, sequentially diversified (typically shotgun) library, a use that is rarely encountered in practice. A typical sequencing client of this service is rather a metagenomic customer who wants to analyze amplicons. However, their sequence diversity is, of course, much lower than in a library obtained by random cleavage of some genomic DNA. This leads to the fact that we are very often far away to meet the technical specifications, especially in amplicon sequencing, and this is not due to some error (detailed analysis below). Therefore, in 2019 we completely eliminated this sequencing setting, because with the vast majority of orders for 2x300 we were unable to meet the manufacturer's specifications. But then, quite understandably, you, our clients, were constantly interested in the possibility of sequencing 2x300, because it just makes sense for those metagenomic libraries, even if the quality or amount of data is lower than what Illumina promised. Therefore, we decided to return this setting to our portfolio of services in 2020, but without any guarantee as to the amount of data and its quality. Below are some of the results you can expect when sequencing at 2x300.
Manufacturer specifications as of today (March 3, 2020 for MiSeq 2x300):

Why are amplicons causing a headache?
In principle, if the templates are amplicons, these specifications can only be met individually. To obtain 44-50 million reads in a single sequencing run with a length of 300 b, where the quality of >70% bases would be higher than 30 (phred score) is in our experience virtually impossible. If we want the highest quality possible, we need to reduce the density of the clusters per sequencing flowcell, so this translates into the amount of data we can deliver. If you insist that you need about 14 Gb of data, as stated by the manufacturer, it will of course be possible, but in the case of the amplicon library you can not count on the quality of reads according to specifications. The density of clusters will have to be so high that they overlap each other, and the quality simply goes down. Not to mention that in order to "disrupt" low sequence diversity, amplicons must be "diluted" with some high diversity sequence (typically PhiX), the presence of which will significantly reduce the amount of data of interest to you.
And what can we do about it?
In a way, we are in part able to influence the sequencing output by adjusting the sequencing run or the nature of the sequencing library to prioritize the amount of data over quality and vice versa. In summary, this is about harmonizing the following parameters on our side:
The given parameters have each their optimum value and if the optimum values are maintained for all parameters, we can always theoretically hope for an optimum result. However, in our experience, the quality of the last few dozen bases from each read will decrease more or less. If the nature of the experiment does not allow the optimal value of any of the parameters, the deviation from the optimal sequencing result is usually rather pronounced, which is typically the case with amplicon libraries.
What you can expect
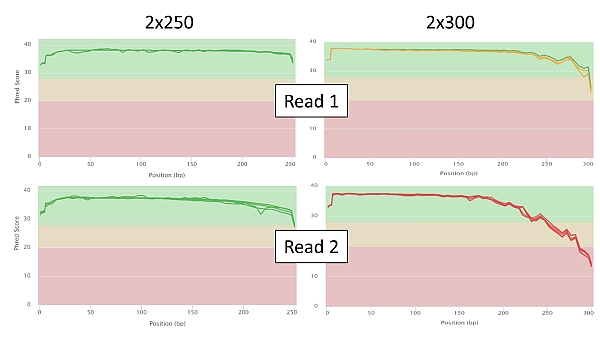
Below are a few selected sequencing quality charts in 2x250 and 2x300 settings for comparison. In the graphs of the Phred score dependence on the position of the particular read base, you can observe a significant decrease in the quality of read in relation to its length as soon as we approach base no. 250 (PE250) or beyond it (PE300). Of course this is more pronounced in read 2.
Summary
For this technology, especially with amplicon libraries, the read length of 2x300 bases is simply far beyond its potential (and probably will remain so) and the results cannot be 100% reliable, which is however the only notable exception for Illumina technology.
In practice, this means that for sequencing at 2x300 b, we do not guarantee the amount or quality of data, regardless of the type of library. However, based on our experience, we are able to provide you with a qualified estimate.
NGS Lab, ngs@seqme.eu